α7烟碱型乙酰胆碱受体的配体化合物及其应用的制作方法
本发明涉及医药技术领域,特别是涉及α7烟碱型乙酰胆碱受体的配体化合物及其应用。
背景技术:
烟碱型乙酰胆碱受体(nachr)是一类门控-递质离子通道,普遍存在于中枢神经系统(cns)和周围神经系统(pns)中,与多种生理功能相关。nachr存在不同的亚型,其一般是由α亚基(如α2-α10)和β亚基(β2-β4)构成。其中,α7烟碱型乙酰胆碱受体(α7nachr)则是由5个完全相同的α亚基构成的同源五聚体,它主要存在于海马,丘脑以及大脑皮质等有关于记忆、学习等的重要区域。近期的临床研究发现,在阿尔茨海默病和帕金森病等神经退行性疾病的病人脑中均发现了α7nachr蛋白质密度的减少,在基因剔除、亚型选择性配体等方面的研究表明:靶向性的α7nachr配体能够提高认知能力以及听觉门控缺陷,例如pnu-282987、pha-543613以及a-582941等高选择性的α7nachr激动剂提高了感觉-门控缺陷、短期工作记忆、以及记忆固化等模型的认知功能。因此,针对α7烟碱型乙酰胆碱受体激动剂及放射性配体的合成也越来越受到广泛的关注。
技术实现要素:
本发明实施例的目的在于提供一种α7烟碱型乙酰胆碱受体的配体化合物,以得到与α7烟碱型乙酰胆碱受体具有高亲和性的放射性或非放射性的配体分子。具体技术方案如下:
一种α7烟碱型乙酰胆碱受体的配体化合物,其具有以下通式中的任一种,
其中,
(1)x为:
(2)r2为氢,且r3为卤素或氨基;或者r3为氢,且r2为卤素或氨基;
(3)r6为氢,且r4和r5结合成:
(4)y为氮或碳;z为
本文中,所说的卤素包括氟、氯、溴或碘。
在一些实施方案中,卤素经过放射性标记或未经放射性标记。
在一些实施方案中,所述卤素为f或18f。
在一些实施方案中,本发明提供的α7烟碱型乙酰胆碱受体的配体化合物可以选自以下化合物:
在一些具体实施方案中,本发明所提供的α7烟碱型乙酰胆碱受体的配体化合物可以作为α7烟碱型乙酰胆碱受体的激动剂。
在一些具体实施方案中,本发明所提供的α7烟碱型乙酰胆碱受体的配体化合物可以作为α7烟碱型乙酰胆碱受体的部分激动剂。
本文所用术语“激动剂”应理解为赋予了其最宽泛的含义,即,作为部分或全部地激活目标材料(例如,α7烟碱型乙酰胆碱受体)至少一种生物活性的任何分子。例如,本发明提供的α7烟碱型乙酰胆碱受体的配体化合物可特异性结合至α7烟碱型乙酰胆碱受体的胞外结构域以诱导胞内信号传递,从而证明其在预防或治疗认知障碍和在神经性康复中的功效。
α7烟碱型乙酰胆碱受体已知在提高例如,学习、记忆和注意力方面的认知功能上很重要。例如,α7烟碱型乙酰胆碱受体与下述疾病有关:轻度认知障碍、阿尔茨海默症、与年龄相关的和其他认知障碍、精神分裂症、注意力缺陷障碍、注意缺陷多动障碍(adhd)、注射或代谢失调引起的痴呆症、路易体痴呆症、抽搐如癫痫、多发性脑梗塞、情绪失调、强迫性和上瘾行为、炎性疾病,以及与控制由这些失调导致的疼痛有关的疾病和病症。α7烟碱型乙酰胆碱受体的活性可通过施用α7受体配体来改变或调节,所述α7受体配体的非限制性实例有:拮抗剂、激动剂、部分激动剂和反向激动剂。α7受体配体可用于治疗和预防这些和各种类型的认知障碍和其他病症和疾病,而其激动剂和部分激动剂已知能在啮齿动物、非人类灵长类和人类中改善认知功能和注意力。
基于此,本发明的另一方面α7烟碱型乙酰胆碱受体的配体化合物在制备预防或治疗认知障碍的药物中的用途。
本文所用术语“认知障碍”是指动物在认知功能或认知领域方面大范围的退化,例如,在工作记忆、注意力和警觉、语言学习和记忆、视觉学习和记忆、推理和解决问题方面,尤其是,例如,在执行能力、任务处理速度和/或社会认知方面。认知障碍已知表现出注意力缺陷、思维紊乱、思维反应迟钝、理解困难、注意力差、失去解决问题能力、记忆不准确、表达思想和/或综合思维以及感觉和行为上有困难或在消除不合理思维上有困难。
术语“治疗”可认为是包括预防、抑制和减缓(消退)动物的与认知障碍相关的疾病、失调或病症,该动物过去从未诊断为患有由认知障碍导致的这类疾病、失调或病症,但其易于患上这类疾病、失调或病症。相应地,术语“治疗上有效量”是指用于缓解、减轻或预防待治疗疾病的症状所需有效剂量的临床指标,或用于降低或延迟这类症状发作的有效活性化合物的有效剂量,其可在待治疗疾病的体内和/或体外模型中通过实验根据经验来确定。
本发明还提供了卤素经过放射性标记的α7烟碱型乙酰胆碱受体的配体化合物,其作为pet(,positronemissioncomputedtomography,正电子发射计算机断层显像)显像剂的用途。
本发明还提供了用于预防或治疗认知障碍的药物组合物,所述组合物包含治疗上有效量的前述的α7烟碱型乙酰胆碱受体的配体化合物;以及药学上可接受的载体。
在一些具体实施方式中,其中所述认知障碍选自下组:早老年性痴呆症、早发性阿尔茨海默病、老年性痴呆症、阿尔茨海默型痴呆症、路易体小体性痴呆症、微小梗塞性痴呆症、aids相关痴呆症、hiv痴呆症、路易体相关痴呆症、唐氏综合征相关痴呆症、皮克氏病、轻度认知功能障碍、与年龄相关的记忆障碍、最近短期记忆障碍、年龄相关认知障碍、药物相关的认知障碍、免疫缺陷综合征相关的认知障碍、血管疾病相关的认知功能障碍、精神分裂症、注意力缺陷障碍、注意缺陷多动障碍(adhd)以及学习缺陷障碍。
本发明所提供的各配体化合物,与α7烟碱型乙酰胆碱受体具有较高的亲和性,是α7烟碱型乙酰胆碱受体的优良配体化合物,进一步地,将本发明提供的α7烟碱型乙酰胆碱受体的配体化合物经过放射性化学标记后,其可以作为pet显像剂,并具有亲和性好、特异性强、选择性高、脑摄取和代谢速率适中的特点,具有临床应用价值。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为[18f]ⅱ-15和ⅱ-15的hplc分析谱图。
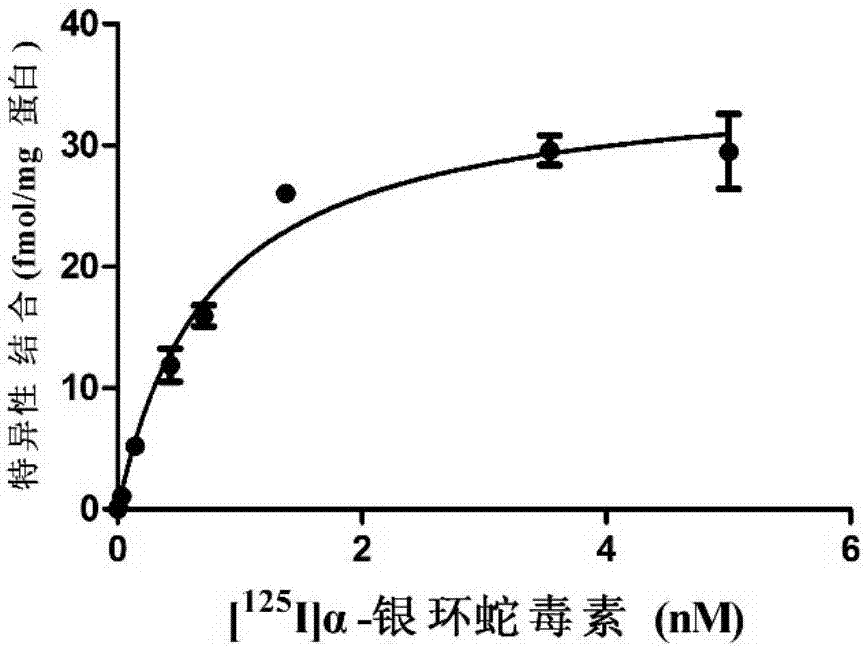
图2为[125i]ɑ-bgt与ɑ7nachrs膜蛋白结合的特异性结合曲线。
图3为[125i]ɑ-bgt与受体膜蛋白结合的hill直线。
图4为scatchard直线;
图5为机率单位y与logd(x)的曲线图。
图6为放射性配体[18f]ⅱ-15在胎牛血清和生理盐水中的稳定性分析的hplc图;
图7为[18f]ⅱ-15的在小鼠脑内的选择性实验结果;
图8为雌性cd-1大鼠pet显像图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1:合成
化合物ⅰ-9的合成路线如下:
(1)化合物ⅰ-2的合成
向洁净干燥的反应瓶中加入化合物ⅰ-1(5g,35.7mmol),用50ml甲醇溶解,置换氮气三次后,将甲醇钠(2.89g,53.5mmol)的甲醇溶液(15ml)滴加到反应体系中,室温下搅拌反应30min;然后将反应体系降温至0℃,逐滴加入溴素(6.3g,39.4mmol),控制反应体系的温度在0-5℃的条件下反应4h;tlc检测反应结束后(ch2cl2:ch3oh=25:1),用少量10%的亚硫酸钠水溶液终止反应,将反应体系降温至-15℃后搅拌析出黄色固体,抽滤之后用冰甲醇(2×5ml)洗涤滤饼,干燥,得黄色固体即为目标产物ⅰ-2(4.2g,53%)。1hnmr(400mhz,dmso)δ7.85(d,j=8.64hz,1h),7.66(d,j=8.64hz,1h);ms(m+h+):m/z=216.97.
(2)化合物ⅰ-3的合成
将化合物ⅰ-2(1g,4.6mmol)加入三口瓶中,用20ml乙醇将其溶解,置换氮气三次后,向其中加入锌粉(1.5g,22.9mmol)和氯化铵(2.46g,46mmol),再次置换氮气;50℃条件下反应16h,tlc检测反应结束后(ch2cl2:ch3oh=25:1),将反应液过滤,收集滤液,减压浓缩后用石油醚/乙酸乙酯(5:1)过柱纯化,得灰黑色固体即为目标产物ⅰ-3(524mg,60%)。1hnmr(400mhz,dmso)δ9.70(s,1h),6.74(d,j=7.8hz,1h),6.49(d,j=7.8hz,1h),5.91(s,1h);ms(m+h+):m/z=188.99.
(3)化合物ⅰ-5的合成
向反应瓶中加入化合物ⅰ-3(0.3g,1.59mmol),用10ml乙醇将其溶解,然后边搅拌边加入二硫化碳(2.42g,31.7mmol)和氢氧化钾(191mg,4.76mmol);80-85℃条件下反应16h,tlc检测反应结束后(ch2cl2:ch3oh=25:1),减压浓缩除去乙醇和二硫化碳,加水溶解,用稀盐酸调节体系的ph至3左右,析出固体,过滤干燥得化合物ⅰ-4粗产品0.26g,直接进行下一步反应。
向三口反应瓶中加入化合物ⅰ-4(0.26g,1.13mmol)和碳酸钾(156mg,1.13mmol)用10mldmf溶解,置换氮气三次后,将反应体系降至0℃;然后将176mg碘甲烷(1.24mmol)逐滴加入到反应瓶中,滴加完毕后在0℃条件下搅拌反应10min,然后室温反应3h;tlc检测反应结束后(ch2cl2:ch3oh=25:1),向反应体系中加入60ml水和20ml乙酸乙酯,搅拌5min后静置;收集有机相,用无水mgso4干燥,过滤,减压浓缩除去溶剂得棕褐色固体ⅰ-5(180mg,65%)。1hnmr(400mhz,dmso)δ8.06(d,j=8.4hz,1h),7.55(d,j=8.4hz,1h),2.79(s,3h);ms(m+h+):m/z=246.93.
(4)化合物ⅰ-6的合成
向反应瓶中依次加入1,4-二氮杂双环[3.2.2]壬烷(200mg,1.58mmol),化合物ⅰ-5(466mg,1.9mmol),三乙胺(321mg,3.17mmol)和10ml异丙醇,边搅拌边加热使体系升温至100℃,待溶剂挥发完后,继续搅拌过夜反应;tlc检测反应结束后(乙酸乙酯/甲醇=10:1),直接用乙酸乙酯(含1%甲醇和1‰三乙胺)作为洗脱剂过柱纯化,得浅黄色固体ⅰ-6(250mg,48.8%)。1hnmr(400mhz,dmso)δ7.68(d,j=8.12hz,1h),7.15(d,j=8.12hz,1h),4.43-4.42(m,1h),3.90(t,j=5.56hz,2h),3.09(t,j=5.76hz,2h),3.04-2.95(m,4h),2.10-2.07(m,2h),1.84-1.76(m,2h);ms(m+h+):m/z=323.905.
(5)化合物ⅰ-9的合成
向反应瓶中加入化合物ⅰ-6(100mg,0.31mmol),化合物ⅰ-7(70mg,0.314mmol),碳酸铯(303mg,0.93mmol)和pd(dppf)cl2(45mg,0.062mmol),将反应体系置换氮气三次后,加入重蒸的1,4-二氧六环(10ml);85-90℃条件下反应,反应结束后减压浓缩除去溶剂,用乙酸乙酯/甲醇(10:1)作为洗脱剂过柱纯化,得淡黄色固体产品即为ⅰ-9(89.4mg,85%)。1hnmr(400mhz,cdcl3)δ8.72(td,j=8.82hz,2hz,1h),8.18(dt,j=1.76hz,4.32hz,1h),7.61(dd,j=8.24hz,1.32hz,1h),7.49(d,j=8.2hz,1h),7.30(td,j=5.94hz,2.04hz,1h),4.61(s,1h),3.99(m,2h),3.21-3.13(m,4h),3.06-2.99(m,2h),2.18-2.16(m,2h),1.88-1.80(m,2h);13cnmr(100mhz,cdcl3)δ163.54,161.83,159.45,158.78,146.72,146.02,141.72,141.05,122.63,122.37,122.01,116.37,114.92,56.96,50.73,46.32,44.33,26.82;ms(m+h+):m/z=340.15.
实施例2:合成
化合物ⅰ-10的合成路线如下:
向反应瓶中加入化合物ⅰ-6(100mg,0.31mmol),化合物ⅰ-8(44mg,0.313mmol),碳酸铯(303mg,0.93mmol)和pd(dppf)cl2(45mg,0.062mmol),将反应体系置换氮气三次后,加入重蒸的1,4-二氧六环(10ml);85-90℃条件下反应,反应结束后减压浓缩除去溶剂,用乙酸乙酯/甲醇(10:1)作为洗脱剂过柱纯化,得浅黄色固体产品ⅰ-10(53mg,50%)。1hnmr(400mhz,cdcl3)δ8.80(d,j=2.32hz,1h),8.52(td,j=7.8hz,2.52hz,1h),7.50(d,j=8.16hz,1h),7.35(d,j=8.12hz,1h),6.99(dd,j=8.52hz,2.8hz,1h),4.65(s,1h),4.03(t,j=5.36hz,2h),3.25-3.19(m,4h),3.10-3.04(m,2h),2.22(m,2h),1.93-1.86(m,2h);13c-nmr(100mhz,cdcl3)δ164.88,163.48,162.49,158.88,149.03,145.90,141.07,139.96,133.43,115.37,112.24,109.51,109.14,55.83,50.57,46.30,26.43,8.70;ms(m+h+):m/z=340.15.
实施例3:合成化合物
化合物ⅱ-5的合成路线如下:
(1)化合物ⅱ-2的合成
向1000ml的反应瓶中加入30.0g9-芴酮(ⅱ-1)(0.17mol)和100ml水,加热至80-85℃,然后在30min内将溴(32.0g,0.2mol)逐滴加入到反应瓶中,滴加完毕后,继续在80-85℃条件下反应4h;待反应体系降至室温后,向其中加入300ml水淬灭反应,然后向其中加入300ml10%的nahso3溶液,搅拌30min后抽滤得到淡黄色固体物质;将该淡黄色固体物质溶于300ml乙醇中,然后在80℃条件下回流搅拌约16h,降至室温后抽滤得淡黄色固体,50℃条件下真空干燥即得2-溴-9-芴酮(ⅱ-2)(39.1g,90.4%)。1hnmr(400mhz,cdcl3)δ7.76(d,j=1.76hz,1h),7.66(d,j=7.32hz,1h),7.61(dd,j=7.88hz,1.76hz,1h),7.51-7.50(m,2h),7.39(d,j=7.88hz,1h),7.34-7.30(m,1h);ms(m+h+):m/z=260.98.
(2)化合物ⅱ-3的合成
向1000ml的反应瓶中加入23.4g2-溴-9-芴酮(ⅱ-2)(0.09mol)和180ml水,加热至80-85℃,然后将180ml65%的硝酸溶液(2.6mol)和180ml98%的硫酸溶液(3.3mol)的混合溶液逐滴加入到反应体系中,大约30min内滴加完毕;将反应体系升温至110-120℃,回流4h,待降至室温后,向其中加入300ml水淬灭反应,过滤得黄色固体;所得固体用水(3×100ml)洗涤后,再加入200ml甲醇打浆洗涤,过滤收集固体,然后在50℃条件下真空干燥,即得2-溴-7-硝基-9-芴酮(ⅱ-3)(21.0g,76.4%)。1hnmr(400mhz,cdcl3)δ8.49(s,1h),8.44(d,j=8.16hz,1h),7.90(s,1h),7.75(d,j=7.92hz,1h),7.71(d,j=8.16hz,1h),7.55(d,j=7.92hz,1h);ms(m+h+):m/z=306.02.
(3)化合物ⅱ-4的合成
向500ml反应瓶中依次加入2-溴-7-硝基-9-芴酮(ⅱ-3)(9.0g,0.03mol),pd2(dba)3(0.6g,0.655mmol),binap(1.3g,2.087mmol),叔丁醇钠(3.3g,0.0343mol),1,4-二氮杂双环[3.2.2]壬烷(3.0g,0.024mol)和1,4-二氧六环(150ml),置换氮气三次后,将反应体系加热至80-85℃,在氮气保护下反应16h;反应结束后,加入150ml乙酸乙酯,然后通过垫有硅藻土的漏斗过滤除去不溶性物质;在滤液中加入10%k2co3溶液调节ph至8.0左右,然后用ch2cl2萃取,直至水相无颜色为止,合并有机相,用无水硫酸钠干燥,过滤,减压浓缩得到蓝紫色产品(ⅱ-4)(2.1g,20.4%),该产品可直接用于下一步反应。1hnmr(400mhz,cdcl3)δ7.56(d,j=7.24hz,1h),7.38(d,j=6.56hz,1h),7.34(d,j=2.8hz,1h),7.14-7.09(m,2h),6.78(d,j=8.16hz,1h),4.13-4.11(m,1h),3.67-3.65(m,1h),3.61-3.58(m,1h),3.15-3.14(m,4h),3.04(m,2h),2.14(m,2h),1.78(m,2h);ms(m+h+):m/z=350.16.
(4)化合物ⅱ-5的合成
向反应瓶中依次加入化合物ⅱ-4(2.1g,6.02mmol),铁粉(1.7g,0.03mol),乙醇(80ml),水(54ml)和浓盐酸(1.8ml),升温至80℃,tlc检测原料反应完全(大约5h);用10%k2co3溶液调节反应体系的ph值至8.0左右,加入150ml乙酸乙酯,通过垫有硅藻土的漏斗过滤,滤饼用乙酸乙酯洗涤,合并有机相,减压浓缩除去溶剂,然后将固体溶解于ch2cl2中,加水洗涤,分离得有机相,用无水na2so4干燥,过滤,减压浓缩得终产物ⅱ-5(1.5g,80%)。1hnmr(400mhz,cdcl3)δ7.12(d,j=8.24hz,1h),7.08(d,j=7.92hz,1h),7.01(d,j=2.44hz,1h),6.88(d,j=2.2hz,1h),6.70(dd,j=8.24hz,2.52hz,1h),6.64(dd,j=7.88hz,2.24hz,1h),4.01(m,2h),3.51-3.48(m,2h),3.13-3.06(m,4h),3.01-2.94(m,2h),2.12-2.05(m,2h),1.75-1.67(m,2h);13cnmr(101mhz,cdcl3)δ194.18,148.14,144.99,135.02,134.79,134.56,132.85,119.00,118.94,117.11,110.32,108.94,55.59,50.91,45.26,43.25,25.50;ms(m+h+):m/z=320.0.
实施例4:合成化合物
化合物ⅱ-6的合成路线如下:
向反应瓶中加入化合物ⅱ-5(1.9g,6mmol)和四氟硼酸(30ml),将反应瓶置于冰浴中,使反应体系降温至0-5℃,然后将亚硝酸钠水溶液(0.5g,7.2mmol,15ml)在15min内逐滴加入到反应体系中;滴加完毕,继续反应30min后,停止搅拌,抽滤,滤饼先用15ml乙醇洗涤,再用15ml甲基叔丁基醚洗涤,最后用甲基叔丁基醚打浆洗涤,过滤得固体;将固体转移至反应瓶中,加热至120℃,tlc检测,当原料完全转换后,将反应体系冷却至室温,加入50mlch2cl2溶解,过滤除去不溶性固体,滤饼用50mlch2cl2打浆洗涤,收集有机相,无水na2so4干燥后,过滤,减压浓缩除去溶剂;粗产品用二氯甲烷和甲醇梯度(ch2cl2:ch3oh=25:1)过柱纯化,得紫色固体即为终产物ⅱ-6(1.2g,67.3%)。1hnmr(400mhz,cdcl3)δ7.24-7.19(m,3h),7.05(d,j=2.28hz,1h),7.02(dd,j=8.4hz,2.4hz,1h),6.74(dd,j=8.32hz,2.56hz,1h),4.0437-4.0391(m,1h),3.55-3.52(m,2h),3.15-3.07(m,4h),3.01-2.94(m,2h),2.12-2.05(m,2h),1.77-1.68(m,2h);13cnmr(101mhz,cdcl3)δ192.51(d,j=2.2hz),162.49(s),160.03(s),148.88(s),140.55(d,j=2.9hz),130.56(s),120.13(s),119.86(s),119.63(s),118.92(s),118.85(s),116.74(s),110.86(s),110.63(s),108.67(s),55.97(s),50.73(s),45.48(s),43.49(s),25.76(s);19fnmr(376mhz,cdcl3)δ-115.21(s);ms(m+h+):m/z=323.2.
实施例5:合成化合物
化合物ⅱ-14的合成路线如下:
(1)化合物ⅱ-8的合成
将三氧化铬(138.0g,1.38mol)溶于120ml水和80ml乙酸的混合溶液中,搅拌使其全部溶解,待用;向反应瓶中加入40.0g荧蒽(ⅱ-7)(0.2mol)和500ml乙酸,将反应体系加热至80-85℃,然后将三氧化铬溶液逐滴加入其中,控制体系的温度在80-85℃,滴加完毕后将反应体系升温至110-120℃,反应2h后,冷却至室温,将反应液倾倒入3l水中,有大量黄色固体析出,抽滤后将固体溶解在600ml2m的naoh溶液中,抽滤除去不溶性杂质,用500ml水洗涤滤饼;加入甲基叔丁基醚洗涤水相,分离得水相,用浓盐酸调节其ph至1.0左右,黄色固体再次析出,抽滤后将其在60℃条件下真空干燥即得目标产物ⅱ-8(9-芴酮-1-羧酸)(29.5g,66.0%)。1hnmr(400mhz,cdcl3)δ8.18(d,j=7.52hz,1h),7.74-7.53(m,5h),7.36(t,j=7.16hz,1h);ms(m+h+):m/z=225.10.
(2)化合物ⅱ-9的合成
向反应瓶中加入9-芴酮-1-羧酸(ⅱ-8)(15.0g,0.067mol)和300ml水,将反应体系加热至80-85℃,然后将10mlbr2(0.23mol)逐滴加入其中,滴加完毕后,继续反应16h,tlc检测(ch2cl2:ch3oh=10:1)反应完全后,向其中加入10%的亚硫酸氢钠水溶液300ml,搅拌30min,过滤得黄色固体,50℃条件下真空干燥得终产物ⅱ-9(19.5g,96.5%)。1hnmr(400mhz,cdcl3)δ8.22(d,j=7.04hz,1h),7.85(s,1h),7.73-7.66(m,3h),7.43(d,j=7.84hz,1h).
(3)化合物ⅱ-10的合成
在搅拌的条件下,向反应瓶中依次加入ⅱ-9(6g,19.87mmol),甲苯(60ml),三乙胺(4.1ml,29.8mmol),dppa(6.4ml,29.8mmol)和叔丁醇(10ml),然后将反应体系升温至110℃回流反应,tlc检测反应结束后(原料,ch2cl2:ch3oh=5:1;中间体,石油醚:乙酸乙酯=5:1),除去溶剂,用硅胶柱纯化(石油醚:乙酸乙酯=30:1)得黄色固体即为产物ⅱ-10(5.5g,74%)。1hnmr(400mhz,cdcl3)δ8.15(d,j=8.56hz,1h),7.71(d,j=1.72hz,1h),7.59(dd,j=7.88hz,1.8hz,1h),7.42(dd,j=7.44hz,8.36hz,1h),7.37(d,j=7.92hz,1h),7.09(d,j=7.16hz,1h),1.55(s,9h).
(4)化合物ⅱ-11的合成
将化合物ⅱ-10(808mg,2.16mmol)溶于40ml乙腈中,向其中再加入22ml1m的盐酸(21.6mmol),82℃条件下加热反应4h,tlc检测反应结束后,冷却至室温,向反应体系中加入适量的1mnaoh溶液调节ph至强碱性,然后用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥,过滤,减压浓缩除去溶剂,得到黄色固体即为产物ⅱ-11(500mg,84.5%)。1hnmr(400mhz,cdcl3)δ7.71(d,j=1.4hz,1h),7.55(dd,j=7.8hz,1h),7.35(d,j=7.9hz,1h),7.22(t,j=7.6hz,1h),6.81(d,j=7.1hz,1h),6.52(d,j=8.4hz,1h),5.54(s,2h);ms(m+h+):m/z=273.99.
(5)化合物ⅱ-12的合成
向25ml的三口烧瓶中加入1.729ml30%的双氧水(16.95mmol),冰浴冷却至0℃后,在氩气保护下,向其中滴加三氟乙酸酐(2.736ml,19.68mmol)的二氯甲烷(2ml)溶液,滴加完毕后,继续在0℃条件下搅拌1.5h(反应混合物由无色变为棕色);然后向反应体系中滴加化合物ⅱ-11(500mg,1.824mmol)的二氯甲烷(4ml)溶液,滴加完毕升至室温,搅拌反应1h,tlc检测反应结束后,加入适量水猝灭反应,用二氯甲烷萃取,收集有机相,用无水硫酸钠干燥,过滤,减压旋蒸除去溶剂,经硅胶柱层析纯化(石油醚:乙酸乙酯=10:1)后,得黄色固体ⅱ-12(212mg,38.2%)。1hnmr(400mhz,cdcl3)δ8.32(d,j=8.4hz,1h),7.75(d,j=1.8hz,1h),7.64(dd,j=7.9hz,1h),7.54(t,j=7.6hz,1h),7.41(d,j=7.9hz,1h),7.31(d,j=7.4hz,1h).
(6)化合物ⅱ-13的合成
将化合物ⅱ-12(100mg,0.329mmol),1,4-二氮杂双环[3.2.2]壬烷(125mg,0.989mmol)和碳酸铯(426mg,1.31mmol)溶于2ml重蒸的无水甲苯中,待用;在氩气保护下,将pd2(dba)3(30mg,0.033mmol)和(±)-binap(61mg,0.099mmol)溶于2ml重蒸的无水甲苯中,90℃条件下搅拌15min后(反应混合物由深紫色浑浊液变为橙黄色澄清液),冷却至室温;然后将前述溶液加入到该反应体系中,氩气保护,60℃条件下搅拌14h后(反应混合物由橙黄色澄清液体变为棕红色液体),冷却至室温,加入10ml水猝灭反应,用二氯甲烷萃取,收集有机相,用无水硫酸钠干燥,过滤,减压旋蒸除去溶剂,经硅胶柱层析纯化(ch2cl2:ch3oh=20:1)后,得紫黑色固体ⅱ-13(48mg,41.8%)。1hnmr(400mhz,cdcl3)δ7.49-7.52(m,2h),7.36(d,j=7.9hz,2h),7.10(s,1h),6.82(t,j=7.6hz,1h),4.13(s,1h),3.62-3.63(m,2h),3.04-3.18(m,6h),2.31(s,2h),1.82-1.83(m,2h);ms(m+h+):m/z=350.37.
(7)化合物ⅱ-14的合成
向反应瓶中依次加入ⅱ-12(2.1g,6.02mmol),铁粉(1.7g,0.03mol),乙醇(80ml),水(54ml)和浓盐酸(1.8ml),升温至80℃反应5h;tlc检测原料反应完全后,用10%k2co3溶液调节ph值至8.0左右,加入150ml乙酸乙酯,通过垫有硅藻土的漏斗过滤,合并有机相,减压浓缩除去溶剂,然后将固体溶解于ch2cl2中,加水洗涤,分离得有机相,用无水na2so4干燥,过滤,减压浓缩得终产物ⅱ-14(1.5g,80%)。1hnmr(400mhz,cdcl3)δ7.30(d,j=8.28hz,1h),7.16-7.12(m,1h),7.06(d,j=2.48hz,1h),6.75(dd,j=8.28hz,2.52hz,1h),6.67(d,j=7.04hz,1h),6.35(d,j=8.28hz,1h),5.43(s,2h),4.10(s,1h),3.58(t,j=5.48hz,2h),3.19-3.13(m,3h),3.07-3.00(m,2h),2.14-2.11(m,2h),1.80-1.74(m,3h);13cnmr(100mhz,cdcl3)δ195.66(s),150.20(s),147.18(s),137.02-136.73(m),136.37(s),131.22(s),121.42(s),116.55(s),115.39(s),108.58(s),108.48(s),57.06(s),51.87(s),46.56(s),44.64(s),26.91(s);ms(m+h+):m/z=320.16.
实施例6:合成化合物
化合物ⅱ-15的合成路线如下:
向反应瓶中加入化合物ⅱ-14(1.3g,4.07mmol)和四氟硼酸(20ml),将其置于冰浴中待温度降至0-5℃后,向其中逐滴加入nano2溶液(0.4g,5.8mmol,10ml),滴加完毕继续反应30min;停止搅拌,抽滤,滤饼先用15ml的乙醇洗涤,再用15ml的甲基叔丁基醚洗涤,最后用甲基叔丁基醚打浆洗涤,过滤得固体;将固体转移至反应瓶中,加热至120℃,tlc检测反应结束后降至室温,加入50ml二氯甲烷溶解,过滤除去不溶性固体,滤饼再用50ml二氯甲烷打浆洗涤,合并有机相,减压浓缩除去溶剂得粗产物,用二氯甲烷和甲醇(25:1)过柱纯化得紫色固体ⅱ-15(446mg,34.0%)。1hnmr(400mhz,cdcl3)δ7.39-7.34(m,1h),7.32(d,j=8.36hz,1h),7.11(d,j=7.36hz,1h),7.09(d,j=2.48hz,1h),6.80-6.74(m,2h),4.10(s,1h),3.60(t,j=5.76hz,2h),3.19-3.12(m,4h),3.05-2.99(m,2h),2.15-2.10(m,2h),1.82-1.73(m,2h);13cnmr(101mhz,cdcl3)δ191.17(s),158.08(s),150.54(s),137.22(s),137.14(s),135.82(s),130.97(s),121.76(s),117.27(s),115.33(s),115.12(s),114.98(s),109.26(s),56.85(s),51.62(s),46.36(s),44.39(s),29.70(s),26.65(s);19fnmr(376mhz,cdcl3)δ-114.11(s);ms(m+h+):m/z=323.2.
实施例7:合成化合物
化合物ⅲ-5的合成路线如下:
(1)化合物ⅲ-2的合成
将5-羟基吲哚(ⅲ-1)(10g,75.1mmol)溶于100ml乙腈中,向该溶液中加入二碳酸二叔丁酯(49.2g,225.3mmol)和dmap(917mg,7.51mmol),室温下搅拌15h;反应结束后,将反应液减压浓缩,加入400ml甲醇溶解,然后再加入k2co3(51.9g,375.5mmol),室温下搅拌4h;反应完全后,用乙酸调节ph至中性,加h2o稀释,萃取有机相(乙酸乙酯作萃取剂),除去溶剂,过柱分离(石油醚:乙酸乙酯=3:1)得产物即为化合物ⅲ-2(12.5g,71%)。1hnmr(400mhz,cdcl3)δ7.99(d,j=6.96hz,1h),7.56(d,j=2.84hz,1h),6.98(d,j=2.44hz,1h),6.85-6.82(m,1h),6.46(d,j=3.64hz,1h),1.66(s,9h);ms(m-h+):m/z=232.11.
(2)化合物ⅲ-3的合成
将化合物ⅲ-2(3.2g,13.7mmol)溶于20mlch2cl2中,二异丙基乙胺(1.77g,13.7mmol)溶于120ml四氢呋喃中,两者混合,然后将混合溶液缓慢加入到三光气(1.3g,4.38mmol)的ch2cl2(100ml)溶液中,室温下搅拌1h后,将1,4-二氮杂双环[3.2.2]壬烷(1.72g,13.7mmol)的ch2cl2溶液缓慢加入其中,室温下反应4h;反应结束后,加h2o稀释,用chcl3萃取,收集有机相,用nacl的饱和水溶液洗涤,无水na2so4干燥,除去溶剂得粗产品;用chcl3和ch3oh(90:10)做展开剂过柱分离得产物ⅲ-3(2.4g,45%)。1hnmr(400mhz,cdcl3)δ8.11(d,j=7.6hz,1h),7.60(s,1h),7.30(m,1h),7.05(d,j=8.96hz,1h),6.52(d,j=3.2hz,1h),4.52-4.41(m,1h),3.89(t,j=5.48hz,1h),3.78(t,j=5.68hz,1h),3.20-3.06(m,6h),2.14-2.11(m,2h),1.82-1.72(m,2h),1.66(s,9h);ms(m+h+):m/z=386.21.
(3)化合物ⅲ-4的合成
将化合物ⅲ-3(2.4g,6.23mmol)溶于20mlch2cl2,待反应液降至0℃后,将10ml三氟乙酸加入其中,然后在30℃条件下搅拌2h;反应结束后,将反应液浓缩,加水稀释,用ch2cl2萃取;水相用饱和nahco3溶液调节ph至8-9,然后用ch2cl2萃取,无水硫酸钠干燥,除去溶剂得产物ⅲ-4(360mg,20%)。1hnmr(400mhz,dmso)δ11.16(s,1h),7.38-7.26(m,3h),6.84(d,j=8.52hz,1h),6.40(s,1h),4.53-4.33(m,1h),3.95(m,1h),3.79(m,1h),3.30(m,6h),2.20-2.10(m,2h),1.95-1.93(m,2h);ms(m+h+):m/z=286.15.
(4)化合物ⅲ-5的合成
0℃条件下,向化合物ⅲ-4(350mg,1.23mmol)的无水dmf(3ml)溶液中加入氢化钠(59mg,1.48mmol),搅拌20min后,将1-氟-2-碘乙烷(321mg,1.85mmol)的无水dmf(3ml)溶液缓慢加入其中,然后在室温下反应2h;反应结束后,向其中加入氯化铵溶液稀释,用乙酸乙酯萃取,收集有机相,用chcl3和ch3oh(95:5,含有20滴氨水)过柱纯化得目标物ⅲ-5(50mg,12.3%)。1hnmr(400mhz,cdcl3)δ7.36-7.34(m,1h),7.26(d,j=8.76hz,1h),7.14(d,j=3.04hz,1h),6.98(d,j=8.76hz,1h),6.48(d,j=3.0hz,1h),4.72(t,j=4.88hz,1h),4.61(t,j=4.88hz,1h),4.51(m,1h),4.39(t,j=4.88hz,1h),4.32(t,j=4.88hz,1h),3.87(t,j=5.72hz,1h),3.76(t,j=5.8hz,1h),3.17-3.01(m,6h),2.13-2.10(m,2h),1.80-1.69(m,2h);ms(m+h+):m/z=332.19.
实施例8:合成化合物
化合物ⅲ-12的合成路线如下:
(1)化合物ⅲ-9的合成
将6-羟基吲哚(ⅲ-8)(10g,75.1mmol)溶于100ml乙腈中,向该溶液中加入二碳酸二叔丁酯(49.2g,225.3mmol)和dmap(917mg,7.51mmol),室温下搅拌15h;反应结束后,将反应液减压浓缩,加入400ml甲醇溶解,然后再加入k2co3(51.9g,375.5mmol),室温下搅拌4h;反应完全后,用ch3cooh调节ph至中性,加h2o稀释,萃取收集有机相(乙酸乙酯作萃取剂);除去溶剂后,过柱分离(石油醚:乙酸乙酯=3:1)得产物ⅲ-9(12.5g,71%)。1hnmr(400mhz,cdcl3)δ7.68(s,1h),7.45(d,j=3.16hz,1h),7.39(d,j=8.4hz,1h),6.79(dd,j=8.4hz,2.2hz,1h),6.48(d,j=3.68hz,1h);ms(m-h+):m/z=232.11.
(2)化合物ⅲ-10的合成
将化合物ⅲ-9(3.2g,13.7mmol)溶于20mlch2cl2中,二异丙基乙胺(1.77g,13.7mmol)溶于120ml四氢呋喃中,两者混合,然后将混合溶液缓慢加入到三光气(1.3g,4.38mmol)的ch2cl2(100ml)溶液中,室温下搅拌1h后,将1,4-二氮杂双环[3.2.2]壬烷(1.72g,13.7mmol)的ch2cl2溶液缓慢加入其中,室温下反应4h;反应完全后,加h2o稀释,用chcl3萃取,收集有机相,用nacl的饱和水溶液洗涤,无水naso4干燥,除去溶剂得粗产品;用chcl3和ch3oh(90:10)做展开剂过柱分离得产物ⅲ-10(2.4g,45%)。1hnmr(400mhz,cdcl3)δ7.97(s,1h),7.54(d,j=3hz,1h),7.50(d,j=8.44hz,1h),7.02(d,j=8.44hz,1h),6.54(d,j=3.64hz,1h),4.50-4.40(m,1h),3.85(t,j=5.64hz,1h),3.77(t,j=5.8hz,1h),3.18-3.02(m,6h),2.13-2.10(m,2h),1.81-1.70(m,2h),1.65(s,9h);ms(m+h+):m/z=386.21.
(3)化合物ⅲ-11的合成
将化合物ⅲ-10(2.4g,6.23mmol)溶于20mlch2cl2,待反应液降至0℃后,将10ml三氟乙酸加入其中,然后在30℃条件下搅拌2h;反应结束后,将反应液浓缩,加水稀释,用ch2cl2萃取;水相用nahco3的饱和溶液调节ph至8-9,然后用ch2cl2萃取,无水naso4干燥,除去溶剂得产物ⅲ-11(360mg,20%)。1hnmr(400mhz,cdcl3)δ8.27(s,1h),7.57(d,j=8.52hz,1h),7.17(d,j=2.36hz,1h),6.87(d,j=8.48hz,1h),6.52(m,1h),4.54-4.44(m,1h),3.90(t,j=5.72hz,1h),3.80(t,j=5.72hz,1h),3.22-3.08(m,1h),2.17-2.13(m,2h),1.84-1.74(m,2h);ms(m+h+):m/z=286.16.
(4)化合物ⅲ-12的合成
0℃条件下,向化合物ⅲ-11(350mg,1.23mmol)的无水dmf(3ml)溶液中加入氢化钠(59mg,1.48mmol),搅拌20min后,将1-氟-2-碘乙烷(321mg,1.85mmol)的无水dmf(3ml)溶液缓慢加入其中,然后在室温下反应2h;反应结束后,向其中加入nh4cl溶液稀释,用乙酸乙酯萃取,收集有机相,用chcl3和ch3oh(95:5,含有20滴氨水)过柱纯化得目标物ⅲ-12(50mg,12.3%)。1hnmr(400mhz,cdcl3)δ7.57(d,j=8.48hz,1h),7.13-7.11(m,2h),6.90-6.87(m,1h),6.51(d,j=3.04hz,1h),4.75(t,j=4.84hz,1h),4.63(t,j=4.92hz,1h),4.51(m,1h),4.38(t,j=4.92hz,1h),4.31(t,j=4.88hz,1h),3.87(t,j=5.64hz,1h),3.77(t,j=5.68hz,1h),3.18-3.01(m,6h),2.13-2.10(m,2h),1.81-1.69(m,2h);ms(m+h+):m/z=332.18.
实施例9:合成化合物
化合物ⅲ-19的合成路线如下:
(1)化合物ⅲ-15的合成
将化合物ⅲ-14(2g,15.85mmol),3-溴吡啶(3g,18.99mmol),pd2(dba)3(320mg,0.35mmol),rac-binap(660mg,1.06mmol)和叔丁醇钠(1.83g,19.04mmol)溶于20ml甲苯中,85℃条件下加热反应16h;反应结束后,冷却,减压浓缩除去溶剂,粗产品用ch2cl2:ch3oh(0.1%nh3·h2o)=10:1过柱分离得棕色固体即为目标产物ⅲ-15(2.2g,68%)。1hnmr(400mhz,cdcl3)δ8.05(d,j=2.4hz,1h),7.98(d,j=4.52hz,1h),7.12-7.09(m,1h),6.90(d,j=8.28hz,1h),3.43-3.39(m,2h),3.23-3.20(m,2h),3.0025-2.9960(m,2h),2.76-2.72(m,2h),2.50-2.47(m,2h),2.34(s,3h);ms(m+h+):m/z=204.15.
(2)化合物ⅲ-16的合成
0℃条件下,将化合物ⅲ-15(2.2g,10.82mmol)溶于80ml乙腈中,然后在30min内把n-溴代丁二酰亚胺(1.93g,10.82mmol)的乙腈(10ml)溶液逐滴加入其中,搅拌30min后升至室温;用40ml水淬灭反应,加入ch2cl2(2×40ml)萃取,收集有机相,用nacl的饱和溶液洗涤,无水naso4干燥,减压浓缩除去溶剂后,粗产品用ch2cl2:ch3oh(0.1%nh3·h2o)=15:1过柱纯化得浅棕色固体即为目标产物ⅲ-16(1.4g,46%)。1hnmr(400mhz,cdcl3)δ7.74(s,1h),7.23(d,j=8.68hz,1h),6.78(d,j=8.68hz,1h),3.43-3.39(m,2h),3.18-3.16(m,2h),3.00(m,2h),2.69-2.66(m,2h),2.54-2.51(m,2h),2.34(s,3h);ms(m+h+):m/z=282.06.
(3)化合物ⅲ-18的合成
将化合物ⅲ-16(1.35g,4.78mmol),化合物ⅲ-17(1.16g,7.21mmol)和pd2(dba)3(553mg,0.48mmol)溶于40ml1,4-二氧六环中,搅拌10min后将10ml碳酸钠(1.93g,18.21mmol)溶液加入其中;将反应体系升温至115℃,回流2h;反应结束后,冷却,向其中加入水/乙酸乙酯(50:50)的混合溶剂;过滤收集固体,用30ml乙酸乙酯洗涤,得浅黄色固体即为目标产物ⅲ-18(1.0g,66%)。1hnmr(400mhz,cdcl3)δ8.23(s,1h),8.16(m,2h),7.81(d,j=8.48hz,1h),7.63(d,j=8.68hz,1h),7.44(d,j=8.52hz,1h),7.22(s,1h),7.03-6.99(m,1h),6.60(s,1h),3.47-3.43(m,2h),3.29-3.27(m,2h),3.01(m,2h),2.79-2.75(m,2h),2.50-2.47(m,2h),2.35(s,3h);ms(m+h+):m/z=319.19.
(4)化合物ⅲ-19的合成
0℃条件下,将94mg氢化钠(2.35mmol)加到化合物ⅲ-18(500mg,1.57mmol)的无水dmf(3ml)溶液中,搅拌30min后,将1-氟-2碘乙烷(546mg,3.14mmol)的无水dmf(3ml)溶液缓慢加入其中,移去冰水浴升至室温,反应2h;反应完全,用nh4cl溶液稀释反应体系,加入ch2cl2萃取,收集有机相,用ch2cl2:ch3oh(0.1%nh3·h2o)=15:1过柱分离即得目标产物ⅲ-19(180mg,31%)。1hnmr(400mhz,cdcl3)δ8.16(s,2h),7.85(d,j=8.6hz,1h),7.64(d,j=8.64hz,1h),7.37(d,j=8.64hz,1h),7.16(d,j=2.8hz,1h),7.03-7.00(m,1h),6.58(d,j=2.88hz,1h),4.80(t,j=4.84hz,1h),4.68(t,j=4.84hz,1h),4.47(t,j=4.92hz,1h),4.40(t,j=4.8hz,1h),3.45-3.41(m,2h),3.31-3.29(m,2h),3.05(m,2h),2.88-2.87(m,2h),2.52-2.50(m,2h),2.40(s,3h);13cnmr(100mhz,cdcl3)δ147.67,143.13,135.99,135.71,131.92,129.22,128.68,121.21,120.68,120.28,118.62,109.19,102.66,83.21,81.53,63.13,54.49,42.37,41.80;ms(m+h+):m/z=365.21.
实施例10:合成化合物
化合物ⅲ-24的合成路线如下:
(1)化合物ⅲ-21的合成
将2.22mldiepa(13.4mmol)和化合物ⅲ-14(847mg,6.71mmol)加入到化合物ⅲ-20(1g,6.71mmol)的正辛醇(20ml)溶液中,120℃条件下过夜反应;反应完全后,减压浓缩除去溶剂,粗产物以ch2cl2:ch3oh(2mnh3·h2o)=4:1为洗脱剂过柱纯化,得白色固体即为目标产物ⅲ-21(1.2g,75%)。1hnmr(400mhz,cdcl3)δ7.165(d,j=9.36hz,1h),6.656(d,j=9.4hz,1h),3.72-3.68(m,2h),3.47(m,2h),3.06(m,2h),2.76-2.72(m,2h),2.59-2.57(m,2h),2.366(s,3h);ms(m+h+):m/z=239.11.
(2)化合物ⅲ-23的合成
室温下,向化合物ⅲ-21(1.1g,4.61mmol)和ⅲ-22(763mg,5.53mmol)的1,4-二氧六环(80ml)混合溶液中加入pd2(dba)3(421mg,0.46mmol)和1,3-双(2,6-二异丙基苯基)氯化咪唑翁(587mg,1.38mmol);置换氮气三次后,向其中加入20%na2co3(10ml,18.4mmol),再次置换氮气(4次);氮气保护,85℃条件下反应19h,反应结束后,冷却;向其中加入200ml乙酸乙酯,通过硅藻土过滤;收集滤液,浓缩除去溶剂,粗产物经过ch2cl2:ch3oh(2mnh3·h2o)=9:1过柱纯化,得棕褐色固体即为目标产物ⅲ-23(850mg,62%)。1hnmr(400mhz,cdcl3)δ7.596(d,j=8.2hz,2h),7.231(m,1h),6.722(d,j=8.24hz,2h),6.388(d,j=9.44hz,1h),3.72-3.70(m,2h),3.68-3.59(m,2h),3.093(m,2h),2.88-2.86(m,2h),2.67-2.63(m,2h),2.407(s,3h);ms(m+h+):m/z=297.16.
(3)化合物ⅲ-24的合成
向1-氟-2-碘乙烷(351mg,2.02mmol)的n,n-二甲基甲酰胺(10ml)溶液中,依次加入k2co3(652mg,4.72mmol)和化合物ⅲ-23(400mg,1.35mmol);室温下搅拌过夜,反应完全后,向其中加入水和乙酸乙酯,分离有机相和水相,水相用乙酸乙酯萃取两次,收集有机相,用无水na2so4干燥,过滤,除去溶剂;粗产品用ch2cl2:ch3oh(2mnh3·h2o)=9:1过柱分离,得目标产物ⅲ-24(160mg,34.6%)。1hnmr(400mhz,cdcl3)δ7.947(d,j=8.68hz,2h),7.587(d,j=9.4hz,1h),7.014(d,j=8.72hz,2h),6.751(d,j=9.4hz,1h),4.844(t,j=3.92hz,1h),4.726(t,j=4.08hz,1h),4.304(t,j=3.96hz,1h),4.234(t,j=4.16hz,1h),3.76-3.72(m,2h),3.60-3.57(m,2h),3.116(m,2h),2.96-2.90(m,2h),2.60-2.58(m,2h),2.431(s,3h);13cnmr(100mhz,cdcl3)δ158.21,148.87,139.36,133.07,121.35,120.55,120.09,118.33,110.13,82.70,81.01,63.01,54.79,42.25,41.70;ms(m+h+):m/z=343.19.
实施例11:合成放射性配体
[18f]ⅱ-15的合成路线如下:
将15mgkryptofix222溶于0.7ml无水乙腈中,2mgk2c2o4溶于0.3ml水中,然后将两者均匀混合配制成1.0mlkryptofix222/k2c2o4淋洗液;用该淋洗液将俘获在qma柱上的18f-淋洗至反应瓶中,100℃条件下用n2流将反应瓶中的溶剂吹干,然后向其中加入0.5ml无水乙腈,再次吹干,该过程反复进行三次,保证充分除去反应瓶中的水分;将标记前体ⅱ-13(2mg)的无水dmso溶液(0.3ml)迅速加入到上述反应瓶中,密封,160℃条件下反应30min;反应结束后,加入蒸馏水(2×10ml)猝灭反应,用注射器吸取反应液通过提前活化的sep-pakc18固相萃取柱;然后,用2ml乙腈将反应产物从该c18小柱上淋洗下来,收集淋洗液,减压浓缩除去溶剂,加入适量的乙腈溶解后以乙腈:水(含有0.2%的乙酸铵)=28:72为流动相进行radio-hplc分离纯化,流速为4ml/min,波长为280nm,半制备柱为
经过radio-hplc分离纯化之后,该放射性配体[18f]ⅱ-15的放化纯度大于98%,放射性标记率约为13.1%(未经衰变校正);将经过纯化的[18f]ⅱ-15和未标记的稳定化合物ⅱ-15共注射进行hplc分析,流动相组成为乙腈:水(含0.2%乙酸铵)=30:70,流速为1ml/min,波长为280nm,分析柱为agelatechnologies,venusilxbpc18(l),5μm,
实施例12:配体化合物的体外竞争实验
(1)饱和结合实验
a)受体蛋白的制备及其浓度的测定
实验过程中用到的受体蛋白均是从雌性sd大鼠(180-200g)的大脑中分离提取而得的。将雌性sd大鼠(180-200g)断颈处死后,迅速取出其大脑置于冰块上,用冰冷的生理盐水冲洗血丝后,解剖出大脑皮层(该区域富集ɑ7nachrs受体蛋白),置于10倍体积冰冷的50mmtris-hcl缓冲溶液(50mmtris,120mmnacl,5mmkcl,2mmcacl2,1mmmgcl2,ph=7.4,4℃)中,然后将烧杯放在冰水浴中用手持式组织匀浆机对该混合物进行匀浆30s(设置为no.6)。将匀浆后的膜溶液分成三等份于50ml离心管中,用低温高速离心机离心20min(4℃,48000g),离心完成后弃去上清液,将下层沉淀物溶于10倍体积冰冷的50mmtris-hcl缓冲溶液中,按照同样的方法对混合物进行匀浆,离心和洗涤。该步骤重复3次之后得到的下层沉淀物即为受体膜蛋白,将其溶于10倍体积冰冷的50mmtris-hcl缓冲溶液中,匀浆使其充分混合均匀。取出10μl混合均匀后的受体膜蛋白溶液,用lowry法测定蛋白的浓度,将剩余的膜溶液分装于2ml离心管中,放在-80℃的冰箱中保存待用。
b)受体膜蛋白的饱和结合实验
饱和结合实验是通过测定放射性配体[125i]ɑ-银环蛇毒素(简称:[125i]ɑ-bgt)与老鼠大脑膜蛋白的结合来进行的。实验中,[125i]ɑ-bgt设置了8个不同的浓度点(0.005-5nm),每个浓度点平行3组。取出保存于-80℃冰箱中的受体膜蛋白,置于4℃条件下冻融,冻融之后根据测定的蛋白浓度加入适当体积冰冷的50mmtris-hcl缓冲溶液(50mmtris,120mmnacl,5mmkcl,2mmcacl2,1mmmgcl2,ph=7.4,4℃)进行稀释。总结合管中反应混合物的总体积为500μl,包括100μl膜蛋白溶液(最终每个试管中蛋白的量为1.5mg),10μl不同浓度的放射性配体[125i]ɑ-bgt和390μl冰冷的50mmtris-hcl缓冲溶液,加样顺序为:膜蛋白,tris-hcl缓冲溶液和[125i]ɑ-bgt(表1)。非特异性结合是通过2μm非标记的ɑ-bgt来确定的,试管中的反应混合物包括100μl膜蛋白溶液(最终每个试管中蛋白的量为1.5mg),10μl不同浓度的放射性配体[125i]ɑ-bgt,100μl2μm的ɑ-bgt和290μl冰冷的50mmtris-hcl缓冲溶液,总体积为500μl,加样顺序为:膜蛋白,tris-hcl缓冲溶液,ɑ-bgt和[125i]ɑ-bgt(表2)。加样结束后,将试管用封口膜封好,涡旋几秒使其充分混匀,然后放在37℃的恒温培养箱中孵育2.5h,孵育完成后,取出试管将其置于冰水浴中以终止受体蛋白与配体的结合,然后用48孔细胞收集器将混合液过滤至whatmangf/b滤纸(提前用0.5%的聚乙酰亚胺溶液浸泡2.5h)上,滤纸用5ml冰冷的50mmtris-hcl缓冲溶液冲洗3次,取下滤纸,剪下滤纸片置于测量的pe管中,用γ-counter测定计数。总结合(tb)与非特异性结合(nsb)之间的差值即为特异性结合(sb),即sb(cpm)=tb(cpm)-nsb(cpm)。
表1饱和结合实验总结合管加样表
表2饱和结合实验非特异性结合管加样表
c)实验结果
饱和结合实验中,放射性配体[125i]ɑ-bgt设立8个不同的浓度(0.005-5nm),每个浓度平行测定3组。实验结果显示,在所测定的浓度范围内,[125i]ɑ-bgt与受体膜蛋白的特异性结合快速达到了饱和,其特异性结合曲线如图2所示;根据特异性结合曲线及公式log[b/(bmax-b)]=nhlog[l]-logkd以log[b/(bmax-b)]对log[l]作图,得到hill直线(如图3所示):y=1.05828x+0.08731(r=0.99031),其斜率即为hill系数nh=1.058,说明[125i]ɑ-bgt与受体膜蛋白的结合为简单的单位点作用系统。
根据单位点作用系统的scatchard方程:b/f=-b/kd+bmax/kd,以特异性结合量b对相应的b/f作图(如图4所示),得到线性回归方程y=46.42857-1.2987x(r=1),根据该方程得到在本实验条件下[125i]ɑ-bgt的平衡解离常数为kd=0.77±0.088nm(95%可信区间为0.36-1.172nm),最大结合量为bmax=35.75±4.64fmol/mg蛋白质(95%可信区间为29.88-41.62fmol/mg蛋白质)。该实验结果与文献中报道的数据相吻合(kd=1.5±0.7nm,bmax=63±17pmol/mg蛋白质),说明的本文中所采用的的测定方法是可信的,可以用于待测化合物生物活性的测定。
(2)竞争结合实验
a)实验方法
为了定量测定配体化合物与ɑ7nachrs的亲和性,我们进行了以[125i]ɑ-bgt为放射性标准品的体外竞争结合实验。实验中,膜蛋白溶液(每个反应管中蛋白的量为1.5mg)与0.4nm的[125i]ɑ-bgt溶液及一系列8个不同浓度(每个浓度平行测定3组)的非标记配体溶液在37℃的恒温培养箱中共同孵育2.5h,孵育完成后,按照与前述饱和结合实验同样的方法处理并用γ-counter测定计数。同时,为了确保该实验体系的准确性和可靠性,以mla(已知的ɑ7nachrs的高选择性,高亲和性配体)为参考配体,测定其与老鼠脑中ɑ7nachrs的亲和性。配体化合物的配制方法和加样方法如下表3和表4所示:
表3配体化合物的配制方法
注:表3只是针对部分浓度范围在10-3-10-10mol/l的化合物的配制方法,对于它化合物在其它浓度范围例如10-6-10-14mol/l、10-5-10-13mol/l,参照图3的方法略作调整即可。
表4竞争结合实验加样表
注:加样顺序为:蛋白,tris-hcl缓冲溶液,药物(包括配体化合物及mla),[125i]ɑ-bgt。
b)实验结果
为了进行比较,选取mla作为参考配体,在相同的实验条件下同时测定mla和所设计的待测化合物对ɑ7nachrs的亲和性。
竞争结合实验中,参考配体mla和一系列待测化合物设立8个不同的浓度(除ⅱ-14为10-6-10-14mol/l、ⅱ-5为10-5-10-13mol/l外,其余化合物为10-3-10-10mol/l),通过抑制0.4nm[125i]ɑ-bgt(kd=0.77±0.088nm)对ɑ7nachrs的结合测定其ic50值,并通过cheng-prusoff公式(ki=ic50/(1+[l]/kd))计算得到各自的ki值。测定结果如表5所示,在该实验条件下,mla的抑制常数ki=2.88±0.78nm,与文献中报道的ki值(1.09±0.09nm)基本相近,表明了本文中所采用的实验方法的可行性。
从表5中可以看出,各配体化合物对ɑ7nachr膜蛋白均显示出亲和性,抑制常数(ki)分布在0.005-450nm范围内,其中化合物ⅱ-14,ⅱ-5,ⅱ-15,ⅱ-6和ⅰ-9对[125i]α-bungaratoxin表现出很强的抑制作用,其ki值分别为0.0069±0.004nm,0.064±0.058nm,2.98±1.41nm,7.24±1.02nm和21.76±1.22nm,说明它们对α7nachr具有很高的亲和性,尤其是化合物ⅱ-14的亲和性(ki=0.0069±0.004nm)已超越了目前国际上同类配体分子的最高值(ki=0.023nm)。
表5mla和各待测配体化合物对ɑ7nachrs的体外结合亲和性(ki,nm)[a]
[a]三次测量平均值±标准偏差
实施例13:体外herg钾离子通道抑制实验
a)实验方法
目前,测定药物对herg钾通道抑制作用的方法主要有三种:全自动膜片钳技术,传统膜片钳技术和fluxortmthalliumassay。本文中采用传统膜片钳技术测定化合物对herg钾通道的抑制作用,该方法是心脏毒性研究的公认标准方法,是最准确的测量方法。实验中,以西沙比利(cisapride,已知对herg钾通道具有强抑制作用的化合物)作为标准化合物,评价待测配体化合物对herg钾通道的抑制作用。
37℃条件下,用dmem培养基(含10%胎牛血清和0.8mg/mlg418)培养herg钾离子通道稳定表达的hek293细胞系(creacell)(5%的co2);经过传代培养后,用trypletmexpress溶液对细胞进行分离,然后将3×103细胞铺到盖玻片上,在24孔板中培养18h后检测细胞的电生理活性,当全细胞记录的herg电流稳定后开始进行药物检测;实验中,每个待测配体化合物设置4个浓度梯度(0.4μμ-50μμ),相邻浓度之间的比例为5,标准化合物西沙比利的浓度范围为1nm-1μμ,相邻浓度之间的比例为10;检测时,每个药物(包括配体化合物和西沙比利)浓度持续5min(或持续作用至电流稳定),然后进入下一个浓度的检测,药物溶液通过重力灌流的方法由低浓度向高浓度依次经过记录室作用于细胞,在记录室中进行液体交换;在进行药物检测之前,设立空白对照组,所有实验组和对照组独立重复检测3次。
b)实验结果
数据处理时,先用空白对照组的记录电流对待测药物每个浓度的作用电流进行校准(待测药物的尾电流峰值/空白对照的尾电流峰值),然后计算待测药物每个浓度对应的抑制率(1-待测药物的尾电流峰值/空白对照的尾电流峰值),求得3次重复实验的平均值和相对标准偏差后,用下列方程计算每个待测化合物的半抑制浓度ic50值:
抑制率=1/[1+(ic50/c)h]
其中,c表示待测药物的浓度,h表示hill系数;曲线的拟合及ic50值的计算通过igor软件辅助完成。
标准化合物西沙比利及待测化合物ⅱ-15,ⅱ-14,ⅱ-6,ⅱ-5对herg钾离子通道的抑制作用结果如表6和7。实验结果显示,化合物ⅱ-14对herg钾离子通道几乎没有抑制作用(ic50>10μm),化合物ⅱ-5对herg钾离子通道具有中度的抑制作用(1μm≤ic50≤10μm),化合物ⅱ-15和ⅱ-6对herg钾离子通道具有较强的抑制作用(0.1μm≤ic50≤1μm),但它们对于该离子通道蛋白的亲和性(ic50在460nm-2500nm范围)远小于其对于α7nachr膜蛋白的亲和性(ic50值在0.21nm-21nm范围)(见表8),此外,虽然该测定方法是研究herg毒性最灵敏的方法,但药物在体内对herg钾电流的抑制作用还与其血液中的浓度等生理因素相关,因此,这些配体分子可以作为潜在的α7nachr激动剂进行进一步的深入研究。
表6标准化合物西沙比利对herg钾电流的抑制作用(ic50,nm)
表7化合物ⅱ-15,ⅱ-14,ⅱ-6,ⅱ-5对herg钾电流的抑制作用(ic50,μm)
表8化合物ⅱ-15,ⅱ-14,ⅱ-6,ⅱ-5对herg钾离子通道及α7nachr的亲和性对比
实施例14:配体化合物ⅱ-15半致死剂量ld50的测定
a)实验方法
取昆明种小鼠(18-20g),雌雄各半共计60只,在实验条件下饲养3天后,禁食不禁水12h,然后对其逐一称重并记录,按体重大小随机分成6组,每组共计10只(雌雄各半);按照预实验探索的浓度范围配制5个不同浓度的溶液和空白对照溶液,实验组相邻浓度之间的比例为0.90-0.95,通过尾静脉注射的给药方式为各组小鼠注射0.1ml药品溶液或空白对照溶液;注射完成后将小鼠分浓度分雌雄放置饲养,并在给药后的0.5h,1h,3h,6h,12h和24h内密切观察并记录小鼠的反应情况(包括行为活动是否活跃,对刺激的反映情况,饮食情况,是否出现抽搐,癫狂,吐血,步履蹒跚等症状),对出现死亡的小鼠要立即将其解剖观察各器官的异常情况;以后每天定时观察,连续观察14天,并在给药后的第2,4,6,8,10,12,14天分别为每组小鼠称量体重并记录;计算在观察期内各组小鼠的死亡率,根据各组的实验浓度和死亡率计算该化合物对小鼠的半致死剂量ld50。
b)实验结果
1.观察期内老鼠的反映情况
实验中,给药后出现死亡的老鼠,在观察阶段内均出现抽搐现象,部分小鼠出现颤抖,吐血,步履蹒跚,狂躁现象;对死亡的小鼠进行解剖,观察其各组织器官,均未发现异常;所有给药的小鼠,在给药后的1h内,均表现出食欲不振现象,后恢复正常;实验组存活的小鼠和空白对照组小鼠在观察期内,体重增长正常,各组小鼠的体重变化没有明显的浓度组差异和雌雄差异。观察期内老鼠的反应情况记录如表9所示:
表9观察期内老鼠的反映情况
2.半致死剂量ld50的计算
ld50的计算方法有很多种,其中由bliss创建又经后来研究者发展的加权概率单位法最精确、严谨,成为研究者公认的标准的ld50计算方法。本文将采取该种计算方法求解待测化合物对小鼠的ld50值。
表10化合物ⅱ-15的ld50计算表
注:权重=权重系数*各组动物数(n);
以机率单位y对logd(x)作图,得到方程y=14.76x-20.53(如图5),当死亡率为50%时,查表得机率单位y为5.00,代入方程计算得x=m=logd=1.73,d=53.70mg/kg。
标准偏差:
根据上述公式,计算得标准偏差sm2=0.0001977577,sm=0.014;
m±1.96sm=1.73±0.02744,即1.70256-1.75744;分别求其反对数,取ld50的95%可信限:50.4150-57.2058mg/kg;因此,化合物ⅱ-15的ld50值为53.70mg/kg。
由上述过程可知,化合物ⅱ-15的半致死剂量ld50值在mg/kg数量级别,远远超出临床上做一次pet显像所注射的剂量,因此,将化合物ⅱ-15用18f标记所得到的放射性配体[18f]ⅱ-15的体内应用是安全的。
实施例15:[18f]ⅱ-15脂水分配系数的测定
大部分神经类药物的研发都必须考虑其穿越血脑屏障(bbb)的能力,药物分子能否穿越血脑屏障与其脂溶性的大小紧密相关。通常情况下,配体分子的脂溶性(logp)数值在1.0-3.0之间时,认为其可以穿越血脑屏障。
在正辛醇与ph=7.4的pbs混合体系中测定[18f]ⅱ-15的脂水分配系数(logp),具体实验方法为,向含有900μlpbs(提前用正辛醇饱和)和1000μl正辛醇(提前用pbs饱和)的10ml离心管中加入100μl10μci放射性配体(生理盐水溶液),将混合溶液涡旋5分钟后,置于离心机上离心5min(7000rmp/min)。离心后分别取100μl有机相和水相于pe试管中,测定放射性计数,并每组再取100μl有机相于10ml离心管中,加入900μl正辛醇和1000μlpbs,按上述涡旋离心,测定放射性计数,如此再重复3次,求平均值。logp=log(有机相计数/水相计数)
通过测定,得放射性配体[18f]ⅱ-15的脂水分配系数logp值为1.64±0.12,该数值符合药物穿越血脑屏障(bbb)进入脑内的脂溶性范围。
实施例16:[18f]ⅱ-15的体外稳定性实验
放射性配体的体外稳定性对其进一步的体内研究具有重要意义,通常情况下,体外稳定性研究是在生理盐水和动物血清中进行的。具体方法是:取10μci经过hplc纯化的放射性配体[18f]ⅱ-15与100μl胎牛血清在37℃条件下分别孵育1h和2h,孵育结束后向其中加入200μl的乙腈使蛋白充分沉淀,然后在4℃条件下离心5min(7000rpm),收集上清液,经滤膜过滤后取100μl进行hplc分析;再取10μci经过hplc纯化的放射性配体[18f]ⅱ-15与100μl生理盐水在室温下分别培养1h和2h,然后直接通过hplc进行分析。
放射性配体[18f]ⅱ-15的体外稳定性实验结果如图6所示,由图6可以看出,[18f]ⅱ-15在生理盐水和胎牛血清中都表现出很好的稳定性。37℃条件下,在胎牛血清中孵育1h(图6中a图)和2h(图6中b图)后,其放化纯均大于98%;室温下,在生理盐水中培养1h(图6中c图)和2h(图6中d图)后,其放化纯仍大于98%。
实施例17:[18f]ⅱ-15的动物体内分布实验
将经过hplc纯化的[18f]ⅱ-15(10μci,溶于0.1ml生理盐水,含5%dmso)通过尾静脉注射的方式注入正常昆明种小鼠体内(18-22g,雌性,n=5),分别在5min,15min,30min,60min,90min时将小鼠断头处死,解剖取出血、脑、心、肺、肝、脾、肾、肌肉、骨和尾,称量各个器官的湿重并用γ-counter测定其计数,每个组织的摄取情况最终以%id/g表示,%id/g=id/g÷1%,其中id/g=组织的放射性计数(counts)÷组织质量(mg),1%=每个时相1%id的平均值-尾部放射性计数/100,该放射性配体的体内分布结果见表11。
表11化合物[18f]ⅱ-15在雌性昆明种小鼠(18-22g)体内的分布情况
表中数据为五次测量的平均值±标准偏差;
由表11可以看出,18f标记的放射性配体[18f]ⅱ-15在小鼠脑内具有非常高的初始脑摄取,在注射5min后其摄取值即达到8.98±0.41%id/g,15min后显示出最高的脑摄取值11.60±0.14%id/g;同时,该放射性配体表现出适宜的脑清除速率,在给药60min、90min后,其脑内的摄取值分别降为5.46±0.27%id/g和3.63±0.25%id/g,这表明了该化合物具有适宜的脑内动力学性质;该结果与目前已报道的进入临床实验的[18f]asem(其最高的脑摄取值出现在给药5min后,为7.5%id/g)相比,已表现出明显的优势。另外,[18f]ⅱ-15在血液中的摄取值很低,表现出很高的脑/血比值,在5min和15min时分别为9.35和9.57。
放射性配体的体内脱氟现象在研究f-18标记的显像剂的过程中是一个必须考虑的问题,从上表的实验结果可以看到,[18f]ⅱ-15的骨吸收值在研究的时间范围内有所增加,但增加速率和增加幅度并不是很大,这表明该化合物的体内脱氟现象比较弱,预计该现象对于放射性配体的体内显像研究并不会产生太大的干扰。此外,在其它的组织器官也有较高的放射性摄取,例如在肾脏和肺部,放射性配体的初始摄取值都很高,但随着时间的延长其摄取值逐渐降低,清除速率很快。该放射性配体与[18f]asem相比,具有更好的脑部吸收特性。
实施例18:[18f]ⅱ-15的脑区域分布实验
将经过hplc纯化的[18f]ⅱ-15(10μci,溶于0.1ml生理盐水,含5%dmso)通过尾静脉注射的方式注入正常昆明种小鼠体内(28-32g,雌性,n=5),分别在给药5min,15min,30min,60min,90min时通过颈椎脱臼的方式将小鼠处死,迅速解剖出脑部置于冰上,用冰冷的生理盐水除去血迹,然后分区域解剖出皮层、纹状体、海马、上下丘、丘脑、小脑和余脑,称量各个脑区域的湿重并用γ-counter测定其放射性计数,每个区域的放射性配体摄取情况最终以%id/g表示,%id/g=id/g÷1%,其中id/g=组织的放射性计数(counts)÷组织质量(mg),1%=每个时相1%id的平均值,该放射性配体的脑区域分布情况见表12。
由表12可以看出,将10μci[18f]ⅱ-15注入小鼠体内后,该放射性配体在α7nachr最为富集的皮层、纹状体和海马区有较高的吸收,并在给药30min后达到峰值,分别为9.39±0.24%id/g,8.37±0.27%id/g和6.31±0.82%id/g,在随后的观察期内,这些区域的摄取值逐渐下降;中度摄取区域为上下丘和丘脑,摄取最低的区域为小脑(小鼠脑内α7nachr分布最少的区域)。该脑区域分布特点与[18f]asem相似,并与文献中报道的α7nachr在体内外的分布情况是一致的,而且其在α7nachr密集区域的摄取值与[18f]asem相比具有一定的优势:[18f]asem在给药后5min时达到吸收的最高值,皮层、纹状体和海马区的摄取值分别为7.2%id/g,6.0%id/g和5.0%id/g。在整个实验过程中,组织/小脑比值逐渐增加,并在给药90min后达到了2.5(皮层),2.9(纹状体)和3.6(海马)(表13),表明了该放射性配体不仅具有较高的脑摄取,而且具有良好的脑区域选择性。
表12化合物[18f]ⅱ-15在雌性昆明种小鼠(28-32g)脑内的分布情况
表13[18f]ⅱ-15在小鼠脑内各组织不同时间点的组织/小脑比值
实施例19:[18f]ⅱ-15的在小鼠脑内的选择性实验
放射性配体对于α4β2nachr的选择性是通过提前5min皮下注射1mg/kg的cytisine(金雀花碱)(0.1ml,以等体积的生理盐水和1,2-丙二醇做溶剂)进行探究的,而对于5-羟色胺受体的选择性是通过提前10min皮下注射2mg/kg的ondanstron(昂丹司琼)(0.1ml,以生理盐水:dmso=5:1(v:v)做溶剂)来进行的。实验中,两组小鼠(n=5,昆明种雌性,28-30g)通过尾静脉注射的方式,注射[18f]ⅱ-150.1ml(60μci),空白对照组小鼠按照相同的方式注射0.1ml对应的溶剂。给药60min后,将小鼠断颈处死,迅速解剖出大脑置于冰上,用冰冷的生理盐水冲尽血迹后,分区域解剖出皮层、纹状体、海马、上下丘、丘脑、小脑和余脑,测定各个区域的质量及放射性计数,最终的摄取情况以%id/g表示。
cytisine是α4β2nachr的选择性激动剂,而ondanstron是5-羟色胺受体的选择性拮抗剂。由图7可以看出,放射性配体[18f]ⅱ-15在实验组和对照组的摄取没有明显差别。这表明,[18f]ⅱ-15对于α4β2nachr和5-羟色胺受体几乎没有结合,该放射性配体对于α7nachr具有良好的选择性。
实施例20:[18f]ⅱ-15的大鼠pet显像
通过尾静脉注射的方式将放射性配体[18f]ⅱ-15(0.3ml,200μci)注射入雌性cd-1大鼠(180-200g)体内,然后用3%的异氟烷将老鼠麻醉至昏迷后,以俯卧的姿势将老鼠固定于小动物micro-pet/ct显像仪上(superarguspet4rl/ct180),扫描显像过程中采用1%的异氟烷维持老鼠处于麻醉状态。分别在给药15min,30min和60min后进行图像采集,观察[18f]ⅱ-15在老鼠脑内的分布情况。
图8分别为雌性cd-1大鼠在注射[18f]ⅱ-1515min、30min和60min后脑部的冠状、矢状、轴状micro-pet显像图。由图8可以看出,[18f]ⅱ-15在老鼠脑内有较高的摄取,其在脑内的分布情况基本与动物体内分布实验结果保持一致,15min时摄取最高,随着时间的延长,放射性配体的浓度逐渐降低,同时,在脑内的滞留较为适宜,在给药60min后依然可以观察到一定浓度的富集。根据上述良好的显像结果,[18f]ⅱ-15适合作为α7nachrpet显像剂。
以上对本发明所提供的α7烟碱型乙酰胆碱受体的配体化合物及其应用进行了详细介绍。本文中应用了具体实施例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其中心思想。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护。
- 还没有人留言评论。精彩留言会获得点赞!