烯烃聚合催化剂组分及其制备方法和烯烃聚合催化剂以及烯烃聚合物的制备方法与流程
本发明涉及烯烃聚合催化剂的领域,具体涉及一种烯烃聚合催化剂组分的制备方法、由该方法制备的烯烃聚合催化剂组分、包含该烯烃聚合催化剂组分的烯烃聚合催化剂以及采用烯烃聚合催化剂的烯烃聚合物的制备方法。
背景技术
ziegler-natta聚烯烃催化剂是以镁、钛、卤素和给电子体作为基本成分的固体颗粒型催化剂,催化剂的颗粒形态对聚合过程中聚合物的粒形起决定性作用,用于调控催化剂颗粒形态主要由三种方法,分别是熔融法、反应法和溶解析出法。其中熔融法和反应法制出的催化剂载体球形度好,表面光滑。但这两种方法均是先制备出球形载体,再由载体制备催化剂,过程繁琐,工艺复杂。如熔融法制备的球型催化剂us4469648、us4399054、wo9844009、us5100849和us4829034和反应法制备球形催化剂专利cn101056894a、us4727051、cn1255436c、cn101190953a。
溶解析出法可以避开载体制备步骤,通过溶解再析出,经载钛等处理直接制备催化剂,是催化剂制备最简单便捷的一种方法,例如cn85100997a、cn1021229c、cn1172966c、cn1763108a、cn103012625a,但是这些方法制备的催化剂的颗粒形态为类球形,粒子表面不光滑,堆积效应强,催化剂分散性能较差。对此类催化剂粒形的改善也是多年来ziegler-natta催化剂研发的难题和热点之一。
技术实现要素:
本发明的目的是为了克服采用现有的溶解析出法制备的ziegler-natta催化剂组分颗粒形态为类球形、粒子表面不光滑、堆积效应强和催化剂组分分散性能较差等缺点,提供一种表面光滑的类球形烯烃聚合催化剂组分及其制备方法和烯烃聚合催化剂以及烯烃聚合物的制备方法。
本发明提供了一种烯烃聚合催化剂组分的制备方法,该方法包括以下步骤:
(1)将卤化镁化合物、醇类化合物和式(i)所示的吡咯烷酮类化合物在溶剂中进行第一接触,形成均匀溶液;
(2)在助析出剂的存在下,将步骤(1)所得的均匀溶液与钛化合物进行第二接触得到混合物;
(3)将步骤(2)所得的混合物与内给电子体化合物进行第三接触,过滤、洗涤和干燥;
其中,r为c1-c20的直链或支链烷基、c3-c20的环烷基、c6-c20的取代或未取代的芳香基或者c2-c20的单脂肪醚基或多脂肪醚基;
r1、r2和r3相同或不同,且各自独立地选自氢、氰基、c1-c10直链烷基和c2-c20单脂肪醚基或多脂肪醚基。
本发明还提供了由上述方法制备的烯烃聚合催化剂组分。
本发明还提供了一种烯烃聚合催化剂,该催化剂含有上述烯烃聚合催化剂组分、烷基铝化合物以及可选的有机硅化合物,其中,所述烯烃聚合催化剂组分中的钛元素与所述烷基铝化合物中的铝元素的摩尔比为1:5-5000,优选为1:20-500。
本发明还提供了一种烯烃聚合物的制备方法,该方法包括:在上述烯烃聚合催化剂的存在下,将烯烃单体进行聚合反应。
本发明和现有技术相比,由于本发明的催化剂组分在采用溶液析出法的制备过程中使用了吡咯烷酮类化合物,能够根本解决颗粒型催化剂分散不均匀、易团聚的问题,制备得到的烯烃聚合催化剂组分的颗粒具有球形结构、表面光滑、颗粒规整且分布窄;并且采用包含该催化剂组分的烯烃聚合催化剂制备的烯烃聚合物的堆积密度提高、流动性能好,改善了在装置上应用时出现堵塞或者物料架桥的问题。
附图说明
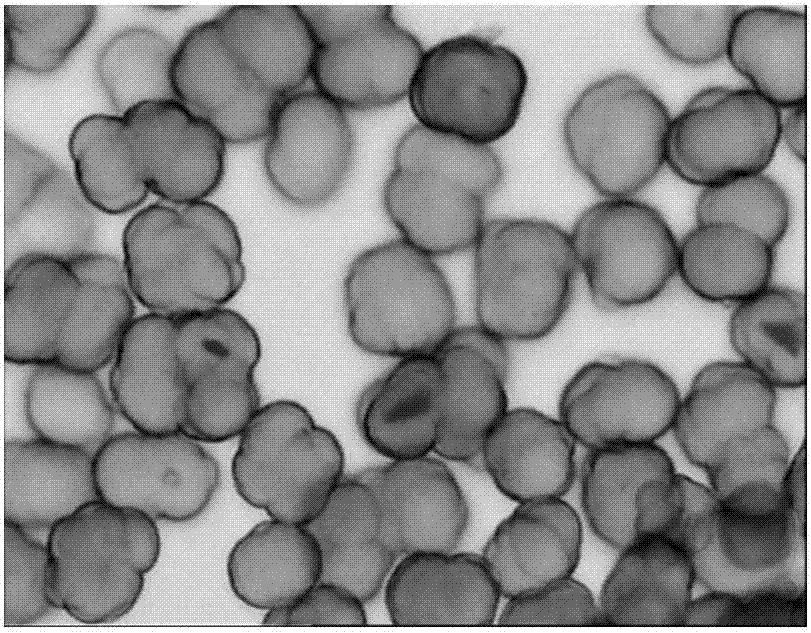
图1是由实施例1制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
图2是由实施例2制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
图3是由实施例3制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
图4是由实施例4制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
图5是由实施例5制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
图6是由对比例1制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
具体实施方式
根据本发明提供的一种烯烃聚合催化剂组分的制备方法,其中,该方法包括以下步骤:
(1)将卤化镁化合物、醇类化合物和式(i)所示的吡咯烷酮类化合物在溶剂中进行第一接触,形成均匀溶液;
(2)在助析出剂的存在下,将步骤(1)所得的均匀溶液与钛化合物进行第二接触得到混合物;
(3)将步骤(2)所得的混合物与内给电子体化合物进行第三接触,过滤、洗涤和干燥;
其中,r为c1-c20的直链或支链烷基、c3-c20的环烷基、c6-c20的取代或未取代的芳香基或者c2-c20的单脂肪醚基或多脂肪醚基;
r1、r2和r3相同或不同,且各自独立地选自氢、氰基、c1-c10直链烷基和c2-c20单脂肪醚基或多脂肪醚基。
在式(i)中,优选地,r为c1-c12的直链或支链烷基、c3-c12的环烷基、c6-c20的取代或未取代的芳香基或者c3-c15的单脂肪醚基或多脂肪醚基。其中,c1-c12的直链或支链烷基例如可以为甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、异戊基、(1,1-二甲基)丙基、(1-乙基)丙基、己基、(1-甲基)戊基、(2-甲基)戊基、(3-甲基)戊基、(4-甲基)戊基、(1-乙基)丁基、(2-乙基)丁基、(1,1-二甲基)丁基、(2,2-二甲基)丁基、(3,3-二甲基)丁基、(1,2-二甲基)丁基、(1,3-二甲基)丁基、(2,3-二甲基)丁基、庚基、辛基、壬基、癸基或十二烷基。c3-c12的环烷基例如可以为环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基、环癸基或环十二烷基。c6-c20的取代或未取代的芳香基例如可以为苯基、间甲基异丙苯基、苄基或9-芴甲基。c3-c15的单脂肪醚基或多脂肪醚基例如可以为二乙基醚基、甲基乙基醚基、二丙基醚基、甲基丙基醚基、二丁基醚基、乙基丁基醚基、二戊基醚基、二己基醚基、二庚基醚基或(2-丁氧基)亚乙基醚基。
在式(i)中,优选地,r1、r2和r3各自独立地选自氢、氰基、c1-c5直链烷基和c3-c12单脂肪醚基或多脂肪醚基。其中,c1-c5直链烷基例如可以为甲基、乙基、丙基、丁基或戊基。c3-c12单脂肪醚基或多脂肪醚基例如可以为二乙基醚基、甲基乙基醚基、二丙基醚基、甲基丙基醚基、二丁基醚基、乙基丁基醚基、二戊基醚基、二己基醚基或(2-丁氧基)亚乙基醚基。
在优选的实施方式中,式(1)所示的吡咯烷酮类化合物选自1-甲氧羰基-3-吡咯烷酮、1-乙氧羰基-3-吡咯烷酮、1-丙氧羰基-3-吡咯烷酮、1-异丙氧羰基-3-吡咯烷酮、1-丁氧羰基-3-吡咯烷酮、1-异丁氧羰基-3-吡咯烷酮、1-叔丁氧羰基-3-吡咯烷酮、1-戊氧羰基-3-吡咯烷酮、1-异戊氧羰基-3-吡咯烷酮、1-(1,1-二甲基)丙氧羰基-3-吡咯烷酮、1-(1-乙基)丙氧羰基-3-吡咯烷酮、1-己氧羰基-3-吡咯烷酮、1-(1-甲基)戊氧羰基-3-吡咯烷酮、1-(2-甲基)戊氧羰基-3-吡咯烷酮、1-(3-甲基)戊氧羰基-3-吡咯烷酮、1-(4-甲基)戊氧羰基-3-吡咯烷酮、1-(1-乙基)丁氧羰基-3-吡咯烷酮、1-(2-乙基)丁氧羰基-3-吡咯烷酮、1-(1,1-二甲基)丁氧羰基-3-吡咯烷酮、1-(2,2-二甲基)丁氧羰基-3-吡咯烷酮、1-(3,3-二甲基)丁氧羰基-3-吡咯烷酮、1-(1,2-二甲基)丁氧羰基-3-吡咯烷酮、1-(1,3-二甲基)丁氧羰基-3-吡咯烷酮、1-(2,3-二甲基)丁氧羰基-3-吡咯烷酮、1-庚氧羰基-3-吡咯烷酮、1-辛氧羰基-3-吡咯烷酮、1-壬氧羰基-3-吡咯烷酮、1-癸氧羰基-3-吡咯烷酮、1-十二烷氧羰基-3-吡咯烷酮、1-苄氧羰基-3-吡咯烷酮、1-9-芴甲氧羰基-3-吡咯烷酮、1-叔丁氧羰基-3-氰基-4-吡咯烷酮、1-叔丁氧羰基-2-甲基-3-吡咯烷酮、1-叔丁氧羰基-2,4-二甲基-3-吡咯烷酮和1-(2-丁氧基)亚乙氧羰基-3-吡咯烷酮中的至少一种,优选为1-乙氧羰基-3-吡咯烷酮、1-叔丁氧羰基-3-吡咯烷酮、1-苄氧羰基-3-吡咯烷酮和1-(2-丁氧基)亚乙氧羰基-3-吡咯烷酮中的至少一种,更优选为1-叔丁氧羰基-3-吡咯烷酮。
在步骤(1)中,相对于1摩尔的以镁元素计的卤化镁,所述吡咯烷酮类化合物用量可以为0.001-0.08摩尔,例如可以为0.001摩尔、0.005摩尔、0.01摩尔、0.02摩尔、0.03摩尔、0.035摩尔、0.041摩尔、0.045摩尔、0.05摩尔、0.055摩尔、0.06摩尔、0.07摩尔和0.08摩尔以及这些点值中的任意两个所构成的范围中的任意值。优选地,相对于1摩尔的以镁元素计的卤化镁,所述吡咯烷酮类化合物用量为0.003-0.05摩尔。
在步骤(1)中,相对于1摩尔的以镁元素计的卤化镁,所述醇类化合物的用量可以为2-4摩尔,例如可以为2摩尔、2.3摩尔、2.5摩尔、2.6摩尔、2.9摩尔、3摩尔、3.2摩尔、3.5摩尔、3.8摩尔和4摩尔以及这些点值中的任意两个所构成的范围中的任意值。优选地,相对于1摩尔的以镁元素计的卤化镁,所述醇类化合物的用量为2.5-3.5摩尔。
在步骤(2)中,相对于1摩尔的以镁元素计的卤化镁,所述助析出剂的用量可以为0.01-1摩尔,例如可以为0.01摩尔、0.05摩尔、0.10摩尔、0.13摩尔、0.15摩尔、0.18摩尔、0.20摩尔、0.25摩尔、0.4摩尔、0.6摩尔和1摩尔以及这些点值中的任意两个所构成的范围中的任意值。优选地,相对于1摩尔的以镁元素计的卤化镁,所述助析出剂的用量为0.05-0.4摩尔。
在步骤(2)中,相对于1摩尔的以镁元素计的卤化镁,所述钛化合物的用量可以为0.5-45摩尔,例如可以为0.5摩尔、1摩尔、5摩尔、8摩尔、10摩尔、20摩尔、25摩尔、30摩尔、31摩尔、35摩尔和45摩尔以及这些点值中的任意两个所构成的范围中的任意值。优选地,相对于1摩尔的以镁元素计的卤化镁,所述钛化合物的用量为8-35摩尔。
在步骤(3)中,相对于1摩尔的以镁元素计的卤化镁,所述内给电子体的用量可以为0.01-5摩尔,例如可以为0.01摩尔、0.05摩尔、0.1摩尔、0.15摩尔、0.2摩尔、0.25摩尔、0.3摩尔、0.5摩尔、0.8摩尔和1摩尔以及这些点值中的任意两个所构成的范围中的任意值。优选地,相对于1摩尔的以镁元素计的卤化镁,所述内给电子体的用量为0.05-1摩尔。
根据本发明,所述卤化镁的通式可以为mgx2,其中,x可以为溴、氯或碘。优选地,所述卤化镁为二氯化镁、二溴化镁和二碘化镁中的至少一种,更优选为二氯化镁。
根据本发明,所述醇类化合物可以为脂肪醇、脂环醇和芳香醇中的至少一种;其中,脂肪醇可以为c1-c10的直链或c1-c10的支链脂肪醇;脂环醇可以为c3-c12的环族脂肪醇;芳香醇可以为c6-c20的芳基醇或c6-c20的烷基芳基醇。优选地,所述醇类化合物为乙醇、丙醇、丁醇、2-乙基己醇、苯甲醇和苯乙醇中的至少一种,更优选为2-乙基己醇。
根据本发明,所述溶剂可以为甲苯、乙苯、苯、二甲苯、氯苯、己烷、庚烷、辛烷和癸烷中的至少一种,优选为癸烷和/或甲苯。
根据本发明,所述助析出剂可以选自有机酸、有机酸酐、醚、酯类和有机酮中的至少一种。优选地,所述助析出剂选自乙酸酐、邻苯二甲酸酐、丁二酸酐、顺丁烯二酸酐、均苯四甲酸二酐、醋酸、丙酸、丁酸、丙烯酸、甲基丙烯酸、丙酮、甲乙酮、二苯酮、甲醚、乙醚、丙醚、丁醚、戊醚、2-乙基-1,3-丙二醇二苯甲酸酯、2-丙基-1,3-丙二醇二苯甲酸酯、邻苯二甲酸二丁酯、邻苯二甲酸二异丁酯和钛酸四丁酯中的至少一种,更优选为邻苯二甲酸酐。
根据本发明,所述钛化合物的通式可以为tixm(or6)4-m,其中,x可以为卤素,r6可以为c1-c20的烃基,m可以为1-4的整数。优选地,所述钛化合物为四氯化钛、四溴化钛、四碘化钛、四丁氧基钛、四乙氧基钛、一氯三乙氧基钛、二氯二乙氧基钛和三氯一乙氧基钛中的至少一种,更优选为四氯化钛。
根据本发明,所述内给电子体可以为本领域常用的各种内给电子体,例如可以选自脂族或芳族羧酸的烷基酯、脂族醚、环脂族醚和脂族酮中的至少一种。优选地,所述内给电子体化合物选自c1-c4饱和脂肪羧酸的c1-c4烷基酯、c7-c8芳香羧酸的c1-c4烷基酯、c2-c6脂肪醚、c3-c4环醚和c3-c6饱和脂肪酮中的至少一种。更优选地,所述内给电子体化合物选自邻苯二甲酸二异丁酯、邻苯二甲酸二正丁酯、邻苯二甲酸二异辛酯、苯二甲酸1,3-二戊酯、甲酸甲酯、甲酸乙酯、甲酸正丙酯、甲酸异丙酯、甲酸丁酯、乙酸甲酯、乙酸乙酯、乙酸正丙酯、乙酸异丙酯、乙酸丁酯、丙酸甲酯、丙酸乙酯、丙酸正丙酯、丙酸异丙酯、丙酸丁酯、丁酸甲酯、丁酸乙酯、丁酸正丙酯、丁酸异丙酯、丁酸丁酯、2-乙基-1,3-丙二醇二苯甲酸酯、2-丙基-1,3-丙二醇二苯甲酸酯、乙醚、丙醚、丁醚、戊醚、己醚、四氢呋喃、丙酮、丁酮、2-戊酮和甲基异丁基酮中的至少一种;进一步优选为邻苯二甲酸二正丁酯和/或邻苯二甲酸二异丁酯。
在步骤(1)中,所述第一接触的条件可以包括:温度为10℃-150℃,优选为30℃-130℃,时间为0.05-10小时,优选为0.1-6小时;
在步骤(2)中,所述第二接触的条件可以包括:温度为-30℃至60℃,优选为-30℃至5℃,时间为0.1-10小时,优选为0.2-8小时;
在步骤(3)中,所述第三接触的条件可以包括:温度为20℃-200℃,优选为30℃-150℃,时间为0.5-8小时,优选为1-6小时。
在步骤(3)中,所述过滤、洗涤、干燥的方法和条件无特殊要求,均可以参照现有技术进行,例如可以用己烷或甲苯将所得固体洗涤、干燥得到烯烃聚合催化剂组分。
根据本发明的一种优选实施方式,本发明提供的制备烯烃聚合的催化剂组分的制备方法包括如下步骤:
(1)将卤化镁与醇类化合物、吡咯烷酮类化合物分散在溶剂中,在10℃-150℃温度下接触0.05-10小时,优选地在30℃-130℃温度下接触0.1-6小时,形成均匀溶液;
(2)在助析出剂存在下,在-30℃至60℃温度下优选为-30℃至5℃温度下,将钛化合物滴入上述均匀溶液或将均匀溶液滴入钛化合物中,接触0.1-10小时,优选为接触0.2-8小时;
(3)再将反应混合物升温至20℃-200℃,优选为30℃-150℃,加入内给电子体化合物,搅拌状态下接触0.5-8小时,优选为1-6小时,滤去母液,再用钛的卤化物及洗涤剂的混合物处理3-4次,处理时间0.5-4小时,滤出液体,用洗涤剂(例如己烷或甲苯)洗涤固体物,干燥后制得烯烃聚合催化剂组分。
本发明提供了由上述的方法制备的烯烃聚合催化剂组分。所述烯烃聚合催化剂组分的颗粒形态具有接近球形结构、粒子表面光滑、颗粒分布较窄。
本发明还提供了一种烯烃聚合催化剂,该催化剂含有所述的烯烃聚合催化剂组分、烷基铝化合物以及可选的有机硅化合物,其中,所述烯烃聚合催化剂组分中的钛元素与所述烷基铝化合物中的铝元素的摩尔比可以为1:5-5000,例如可以为1:5、1:20、1:100、1:100、1:200、1:300、1:400、1:450、1:500、1:1000、1:2000、1:5000以及这些点值中的任意两个所构成的范围中的任意值,优选为1:20-500。
根据本发明,所述烷基铝化合物通式可以为alr'n'x'3-n'所示的化合物,其中,r'可以为氢、c1-c20的烷基或c6-c20的芳基,x'可以为卤素,n'可以为1-3的整数。优选地,所述烷基铝化合物为三甲基铝、三乙基铝、三异丁基铝、三辛基铝、一氢二乙基铝、一氢二异丁基铝、倍半乙基氯化铝和二氯乙基铝中的至少一种,最优选为三乙基铝。
根据本发明,作为外给电子体的所述有机硅化合物的通式可以为rn”si(ory)4-n”,n”可以为0-3的整数,r可以为烷基、环烷基、芳基、卤化烷基、卤素和氢原子中的一种或多种,ry可以为烷基、环烷基、芳基和卤化烷基中的至少一种。优选地,所述有机硅化合物为三甲基甲氧基硅烷、三甲基乙氧基硅烷、三甲基苯氧基硅烷、二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、甲基叔丁基二甲氧基硅烷、二苯基二甲氧基硅烷、二苯基二乙氧基硅烷、二环已基二甲氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、乙烯基三甲氧基硅烷、甲基环己基二甲氧基硅烷、二环戊基二甲氧基硅烷、2-乙基哌啶基-2-叔丁基二甲氧基硅烷、(1,1,1-三氟-2-丙基)-2-乙基哌啶基二甲氧基硅烷和(1,1,1-三氟-2-丙基)-甲基二甲氧基硅烷中的至少一种,最优选为甲基环己基二甲氧基硅烷。
本发明还提供了一种烯烃聚合物的制备方法,该方法包括:在本发明提供的所述烯烃聚合催化剂的存在下,将烯烃单体进行聚合反应。
根据本发明,所述聚合物的聚合方法有如下两种:第一,将一种或多种烯烃与上述的烯烃聚合催化剂和烷基铝化合物接触,其中,上述一种或多种烯烃中乙烯摩尔含量在80%以上;第二,将一种或多种烯烃与上述的烯烃聚合催化剂、烷基铝化合物和外给电子体有机硅化合物接触。在第二种聚合方法中,对上述烯烃没有限制。
对于主要用于乙烯的聚合反应,当烯烃单体中仅仅是部分(小于20摩尔%)除乙烯以外的烯烃时,优选采取上述第一种方式接触(不需要使用外给电子体);当烯烃单体中的乙烯含量在80摩尔%以下时,优选采用上述第二种方式接触。
根据本发明,所述烯烃可以为各种常用的烯烃,例如可以为c2-c6的1-烯烃中的至少一种,优选为乙烯、丙烯、1-正丁烯、1-正戊烯、1-正己烯、1-正辛烯和4-甲基-1-戊烯中的至少一种。
根据本发明,所述烯烃聚合条件可以为本领域常用的烯烃聚合条件,温度为0℃-150℃,时间为0.5-5小时,压力为0.1-10mpa。在本发明中,所述压力是指表压。优选情况下,本发明的烯烃聚合方法在溶剂存在下进行,以烯烃聚合催化剂组分中的钛元素计,上述烯烃聚合催化剂在溶剂中的浓度可以为本领域常规浓度,例如可以为0.0001-1摩尔/升,优选为0.0005-0.75摩尔/升。优选地,上述烯烃聚合在氢气存在下进行,氢气的加入量可以为本领域常规用量,优选为0.01-20升(标准状态下),更优选为0.1-18升(标准状态下)。
以下结合具体实施例对本发明进行详细描述,但并不限制本发明。
以下实施例中,涉及的测试方法如下:
1、催化剂的收率:催化剂的收率%=所得催化剂质量/所用氯化镁质量×100%。
2、催化剂中的钛含量:采用紫外-可见分光光度计721型进行测定。
3、催化剂中酯含量测定:采用色谱法,用催化剂干粉经稀酸分解后,用萃取剂萃取其中的内给电子体化合物,用agilent6890n气相色谱仪进行测定。
4、催化剂的平均粒径(d10、d50、d90)及粒径分布值(span=(d90-d10)/d50)采用masterssizer2000粒度仪(由malverninstrumentsltd制造)测定。
5、聚合物熔融指数(mi)的测定:采用意大利ceast公司的6932型熔体流动指数测定仪进行测定,参照gb/t3682-2000标准。
6、聚合物堆积密度(bd):用单位容积中松散固体重量的方法来测定,参照astmd1895-96标准。
7、聚合物等规度(ii)采用庚烷抽提法测定:2克干燥的聚合物样品,放在抽提器中用沸腾庚烷抽提6小时后,将剩余物干燥至恒重,所得的聚合物重量(g)与2(g)的比值即为等规度。
8、聚合物细粉采用振动筛筛分称重方法计算所得。
实施例1-6用于说明本发明的烯烃聚合催化剂组分、制备方法、烯烃聚合催化剂和烯烃聚合方法。
实施例1
催化剂组分的制备:在经过高纯氮气重复置换的反应釜中,依次加入0.0525摩尔无水氯化镁、22ml癸烷、0.154摩尔2-乙基己醇和0.00184摩尔n-叔丁氧羰基-3-吡咯烷酮,在搅拌转速450rpm、温度为130℃的条件下,反应2.0小时,然后加入0.0081摩尔邻苯二甲酸酐,继续反应一小时,冷却至室温,得到稳定均匀的醇合物溶液。
将上述制备的醇合物均匀溶液加到经氮气充分置换、装有-20℃的1.28摩尔四氯化钛的反应器中,通过搅拌使它们在低温下充分接触,经4小时后,升温至110℃,加入0.0129摩尔邻苯二甲酸二异丁酯,反应2小时,反应结束后,过滤出液体,再加入1.64摩尔四氯化钛,在110℃下,继续反应2小时,反应结束后,过滤出液体,用120ml己烷洗涤5次,干燥,制得固体钛催化剂组份。
图1示出了由实施例1制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
丙烯聚合实施例1a:在一个5升高压釜中,经气相丙烯充分置换后,在室温下加入5毫升三乙基铝的己烷溶液(三乙基铝的浓度为0.5毫摩尔/毫升)、l毫升甲基环己基二甲氧基硅烷(chmms)的己烷溶液(chmms的浓度为0.1毫摩尔/毫升)、10毫升无水己烷和10毫克实施例1制备的固体催化剂组分。关闭高压釜,引入1升标准状态下的氢气和1.15千克的液体丙烯;在搅拌下10分钟内将温度升至70℃。在70℃下聚合反应1小时,反应结束后停搅拌,除去未聚合的丙烯单体,收集聚合物,称重计算催化剂活性(ac)。具体结果详见表1和表2。
实施例2
催化剂组分的制备:方法同实施例1,不同的是使用0.00153摩尔n-乙氧羰基-3-吡咯烷酮代替0.00184摩尔n-叔丁氧羰基-3-吡咯烷酮。
图2示出了由实施例2制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
丙烯聚合实施例2a:方法同实施例1a,使用实施例2制备的催化剂,具体结果详见表1和表2。
实施例3
催化剂组分的制备:方法同实施例1,不同的是使用0.00217摩尔n-苄氧羰基-3-吡咯烷酮代替0.00184摩尔n-叔丁氧羰基-3-吡咯烷酮。
图3示出了由实施例3制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
丙烯聚合实施例3a:方法同实施例1a,使用实施例3制备的催化剂,具体结果详见表1和表2。
实施例4
催化剂组分的制备:方法同实施例1,不同的是使用0.00163摩尔n-(9芴甲)氧羰基-3-吡咯烷酮代替0.00184摩尔n-叔丁氧羰基-3-吡咯烷酮。
图4示出了由实施例4制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
丙烯聚合实施例4a:方法同实施例1a,使用实施例4制备的催化剂,具体结果详见表1和表2。
实施例5
催化剂组分的制备:方法同实施例1,不同的是使用0.00234摩尔n-(2-丁氧基)亚乙氧羰基-3-吡咯烷酮代替0.00184摩尔n-叔丁氧羰基-3-吡咯烷酮。
图5示出了由实施例5制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
丙烯聚合实施例5a:方法同实施例1a,使用实施例5制备的催化剂,具体结果详见表1和表2。
实施例6
催化剂组分的制备:在经过高纯氮气重复置换的反应釜中,依次加入0.0317摩尔无水氯化镁、30ml甲苯、0.03摩尔丁醇、0.08摩尔2-乙基己醇和0.000951摩尔n-叔丁氧羰基-3-吡咯烷酮,在搅拌转速400rpm、温度为70℃的条件下,反应5.0小时,然后加入0.0015摩尔2-乙基-1,3-丙二醇二苯甲酸酯,继续反应一小时,冷却至室温,得到稳定均匀的醇合物溶液。
将上述制备的醇合物均匀溶液加到经氮气充分置换、装有0℃的1.28摩尔四氯化钛的反应器中,通过搅拌使它们在低温下充分接触,经4小时后,升温至60℃,加入0.0254摩尔苯甲酸乙酯,反应1小时,反应结束后,过滤出液体,再加入0.317摩尔四氯化钛,在60℃下,继续反应1小时,反应结束后,过滤出液体,用140ml己烷洗涤5次,干燥,制得固体钛催化剂组份。
丙烯聚合实施例6a:方法同实施例1a,使用实施例6制备的催化剂,具体结果详见表1和表2。
对比例1
催化剂组分的制备:方法同实施例1,不同的是在溶解过程中不加入n-叔丁氧羰基-3-吡咯烷酮。
图6示出了由对比例1制备的烯烃聚合催化剂组分在光学显微镜下放大1600倍的颗粒形态照片。
丙烯聚合对比例1a:方法同实施例1a,使用对比例1制备的催化剂,具体结果详见表1和表2。
表1
表2
从表1、表2和图1-6可以看出,本发明通过引入吡咯烷酮类物质制备得到的催化剂的性能优异,颗粒形态明显改善,表面光滑,催化剂颗粒分布窄。同时聚合得到的聚合物的堆积密度提高,流动性能好。
- 还没有人留言评论。精彩留言会获得点赞!