胺基富勒烯衍生物及其制备方法和应用与流程
本发明属于生物医药材料领域。具体地,本发明涉及一种胺基富勒烯衍生物,其制备方法和其在生物医学领域方面的应用,更具体的,该应用涉及胺基富勒烯衍生物在清除自由基,以及保护细胞对抗自由基损伤的应用。
背景技术
自由基指外层轨道含有未配对电子的基团,具有高度的化学活性。
生物体内不可避免的产生自由基,在正常的生命过程中,低水平的自由基为维持生命所必需。体内自由基不断产生,也不断地被清除,两者处于动态平衡之中,使之维持在一个正常的生理水平上。研究表明:一定浓度的自由基是机体进行正常生命活动的必要条件之一,但过多的自由基对机体是有害的。它可以直接攻击dna,造成其永久性损伤;可以攻击生物膜的不饱和脂肪酸引起脂质过氧化反应;可以攻击蛋白质引起其结构与构象的改变,造成肽链断裂、聚合与交联。所有这些生物大分子结构与功能的改变,必然会导致细胞功能的紊乱,从而导致许多疾病,如冠心病、衰老、脑损伤、炎症、肺损伤、眼组织损伤、动脉粥样硬化、神经退行性疾病、风湿性关节炎和其他自身免疫疾病。
人体内的自由基主要来源于两方面,一是外源性自由基,包括由大气污染物质(no2),电离辐射(紫外线),某些药物(抗肿瘤药物,抗生素,解热镇痛药),各种微量元素(铅,铝等),酒精,高压氧中毒引起;二是内源性自由基,主要是通过某些酶促反应或非酶促反应而产生的。
现在已得到认可的生物抗氧化的物质或外源性天然自由基清除剂主要包括:维生素类(维生素e、维生素c和胡萝卜素),黄酮类化合物(黄酮、菇类和多糖类),木质素类,生物碱,微量元素、金或钯纳米颗粒以及氧化铈纳米颗粒,而这些物质所能清除的自由基比较单一,捕获自由基的效率低,用量大,并且有副作用。
富勒烯是由不同数目的碳原子组成的具有封闭结构的原子团簇。富勒烯由于具有稳定性好、强度高、比表面积大等优点,在生物医药、储能、贮氢、催化等方面的应用中都表现出优异的性能。并且富勒烯的缺电子多烯结构,容易与自由基发生加成反应,被称为“自由基的海绵”,可保护细胞免受自由基伤害。然而富勒烯本体只能溶于部分有机溶剂,无法溶于水,因此不能直接应用于生物体系。
目前已发展出了多种水溶性富勒烯衍生物,而制备水溶性富勒烯衍生物的方法主要有两大类:通过聚合物对富勒烯分子进行包覆;或者将水溶性基团如氨基、羧基通过化学反应键合到富勒烯碳笼上。但是,聚合物包覆法存在富勒烯泄露的潜在危险,且包覆后的颗粒较大,进入体内后很难被细胞吞噬,限制了采用这种方法制备的水溶性富勒烯衍生物的体内应用。而化学键和法多需要将富勒烯溶解在有机溶剂中进行反应,富勒烯在有机溶剂中的溶解度有限,因此难以大规模量产,并且后期纯化去除有机溶剂的过程繁琐而去除效率低。另外,目前化学键合法是固液法,水溶性基团是键和在富勒烯分子聚集的纳米团簇上,得到的富勒烯衍生物是纳米颗粒,易团聚,水溶性仍较差,冷冻干燥后的产物不溶于水。此外,在水溶性富勒烯衍生物中,富勒醇的多羟基会破坏碳笼的完整性,导致其清除自由基的效果较本体有所下降,并且富勒醇不适合进一步衍生化,限制了其在生物医学领域的其他应用。羧酸富勒烯衍生物的制备过程繁琐、键合的羧酸数量少、水溶性较差。
因此,如何对富勒烯进行修饰改性,制备水溶性的富勒烯衍生物,同时保持甚至提高其自由基清除能力,以满足生物医药应用的需求,仍然是需要解决的技术问题。
此外,如何改进水溶性富勒烯衍生物的制备方法,使其能方便地通过调节反应温度或反应时间,来控制获得的水溶性富勒烯衍生物上键合的修饰基团的数量,并且,能改善所制备的水溶性富勒烯衍生物的水溶性、粒径以及分散性,以解决目前所制备获得的水溶性富勒烯衍生物易团聚、水溶性不高等问题。
技术实现要素:
本发明提供了一种制备胺基富勒烯衍生物的新方法,所述方法制备的胺基富勒烯衍生物的水溶性高、粒径小、不易团聚,具有很高的清除自由基的活性。
本发明的一个方面是提供一种胺基富勒烯衍生物。
根据本发明,所述胺基富勒烯衍生物的分子式为c2n(有机胺)m,其中,c2n代表富勒烯,30≤n≤60,有机胺为10℃-40℃下为液体的有机胺,5≤m≤15,有机胺分子键合在富勒烯表面。
根据本发明,优选30≤n≤45,例如n为30、35、38、39、42等。
根据本发明,所述富勒烯优选为c60、c70、c76、c78或c84。在本发明的一个实施方式中,所述富勒烯为c60或c70。
根据本发明,所述有机胺优选为乙二胺、丙二胺、丙醇胺和二乙醇胺中的至少一种。在本发明的一个实施方式中,所述有机胺为乙二胺。
本发明的第二个方面是提供一种胺基富勒烯衍生物的制备方法。
根据本发明,所述制备方法包括如下步骤:
(a)将富勒烯溶解在有机溶剂中,向其中加入有机胺进行反应,待出现沉淀后,收集沉淀物;
(b)将步骤(a)得到的沉淀物加入有机胺中进行反应;
(c)将步骤(b)得到的反应溶液干燥,用水溶解所述干燥后的剩余固体,得到相应的水溶液。
根据本发明,所述有机胺为10℃-40℃下为液体的有机胺。在本发明的一个实施方式中,所述有机胺为乙二胺、丙二胺、丙醇胺和二乙醇胺中的至少一种。在本发明的一个实施方式中,所述有机胺为乙二胺。
根据本发明,所述富勒烯的分子式为c2n,30≤n≤60,优选30≤n≤45。在本发明的一个实施方式中,所述富勒烯为c60、c70、c76、c78或c84。在本发明的一个实施方式中,所述富勒烯为c60或c70。
在本发明的一个实施方式中,所述胺基富勒烯衍生物的分子式为c2n(有机胺)m,其中,c2n代表富勒烯,30≤n≤60,有机胺为10℃-40℃下为液体的有机胺,5≤m≤15,有机胺分子键合在富勒烯表面。
优选30≤n≤45,例如n为30、35、38、39、42等。
优选所述富勒烯为c60、c70、c76、c78或c84。在本发明的一个实施方式中,所述富勒烯为c60或c70。
优选所述有机胺为乙二胺、丙二胺、丙醇胺和二乙醇胺中的至少一种。在本发明的一个实施方式中,所述有机胺为乙二胺。
根据本发明,步骤(a)中的有机溶剂为能溶解富勒烯的有机溶剂,包括但不限于:邻二甲苯、甲苯、二甲苯、二硫化碳和氯苯中的至少一种。在本发明的一个实施方式中,所述有机溶剂为邻二甲苯。
采用能溶解富勒烯的有机溶剂将富勒烯溶解,有利于液态有机胺与溶解在有机溶剂中的富勒烯发生反应,使步骤(a)的反应中,有机胺更大几率和比例的是键合到单一富勒烯分子上,而不是键合到富勒烯分子的纳米团簇上,有利于增强键合了有机胺的单一富勒烯分子的极性,使其容易从有机溶剂中析出。即,从有机溶剂中形成的沉淀主要是键合了有机胺的单一富勒烯分子,键合了有机胺的富勒烯分子纳米团簇的比例较小,从而奠定本发明制备方法的可控性,以及奠定本发明制备方法能提高所制备的衍生物的水溶性的特点。
根据本发明,步骤(a)中将富勒烯溶解到有机溶剂中的方法,可以采用本领域已知的各种方法,包括但不限于:超声、搅拌或震荡等。
根据本发明,步骤(a)中富勒烯与有机胺的质量比可以为1:(100~300),优选为1:(150~200),例如,富勒烯与有机胺的质量比为1:180,1:160,1:170,1:190等。
根据本发明,步骤(a)中的反应时间可以为几分钟到数小时,可根据反应的温度进行调节。例如,如果在室温下反应,搅拌所述反应体系反应1小时;或者,在加热搅拌回流条件下反应十几分钟。根据本发明,加热搅拌的温度可以根据有机胺的沸点而定。根据本发明,搅拌速率可根据情况调整,例如可以为1000rpm/min。
根据本发明,步骤(b)中富勒烯与有机胺的质量比可以为1:(500~1500),优选为1:(800~1200),更优选为1:(900~1100),例如,富勒烯与有机胺的质量比为1:1080,1:1000,1:900等。
根据本发明,步骤(b)中反应的时间可以为数小时到数十小时,可以根据反应的温度进行调节,例如可以是2~24小时。根据本发明的具体实施例,可以在室温下,搅拌所述混合溶液反应24小时;或者,在加热搅拌回流条件下反应8小时。根据本发明,加热搅拌的温度可以根据有机胺的沸点而定。根据本发明,搅拌速率可根据情况调整,例如可以为1000rpm/min。
根据本发明,步骤(b)的反应中,有机胺分子进一步键合到富勒烯分子上,形成键合了多个有机胺分子的富勒烯分子,所述富勒烯分子的极性进一步增大,从而能完全溶解于有机胺中,反应最终能得到澄清透明的溶液。
根据本发明,步骤(a)和(b)的反应优选在惰性气体气氛下进行,例如氮气、氦气或氩气气氛下。
根据本发明,步骤(c)中的干燥处理,可采用本领域已知的各种干燥手段,包括但不限于:旋转蒸发仪、冷冻干燥等。在本发明的一个实施方式中,采用旋转蒸发仪将有机胺蒸发除去。
根据本发明,步骤(c)中用水溶解所述干燥后的剩余固体时,如果仍有不溶物,可以通过调节溶液的ph值为4~6,促进不溶物溶解。在本发明的一个实施方式中,通过加入酸溶液调节所述溶液的ph值,所述酸溶液为0.5~1.5mol/l的酸溶液。所述酸可以是本领域已知的各种无机酸和有机酸,例如盐酸、硫酸、醋酸、磷酸等。
根据本发明,步骤(c)中如果加入酸溶液后,仍有不溶物,该不溶物一般就是未键合有机胺的富勒烯分子或者仅键合少量有机胺的富勒烯分子。所述不溶物可通过常规的过滤手段除去。通过步骤(c),可进一步提高产物中键合多个有机胺分子的富勒烯分子的比例。
根据本发明,在所述的步骤(c)后任选增加提纯操作。
根据本发明,所述的提纯操作包括:
步骤(d):过滤步骤(c)得到的水溶液,调节过滤所得水溶液的ph值为4~6,透析至所述水溶液的电导率小于1μs/cm。
任选,进一步包括步骤(e):将步骤(d)透析后的溶液进行阴离子交换处理。如果步骤(c)在溶解干燥后的剩余固体时,加入了酸溶液,则通过阴离子交换处理操作,能将胺基富勒烯酸式盐转换为胺基富勒烯。
任选,进一步包括步骤(f):将步骤(e)阴离子交换处理后的溶液进行透析,至所述溶液的电导率小于1μs/cm。
根据本发明,所述步骤(d)能透析去除未反应的有机胺。
根据本发明,所述步骤(f)能透析去除残留的无机物。
在本发明的一个具体实施方式中,所述透析处理可以采用透析袋,例如截止分子量为3500的透析袋。
在本发明的一个具体实施方式中,所述透析处理利用电导率不低于18.2mωcm的超纯水进行透析。
本发明的第三个方面是提供由本发明第二个方面所述的制备方法制备获得的胺基富勒烯衍生物。
优选,所述胺基富勒烯衍生物的分子式为c2n(有机胺)m,其中,c2n代表富勒烯,30≤n≤60,有机胺为10℃-40℃下为液体的有机胺,5≤m≤15,有机胺分子键合在富勒烯表面。
优选30≤n≤45,例如n为30、35、38、39、42等。
优选所述富勒烯为c60、c70、c76、c78或c84。在本发明的一个实施方式中,所述富勒烯为c60或c70。
优选所述有机胺为乙二胺、丙二胺、丙醇胺和二乙醇胺中的至少一种。在本发明的一个实施方式中,所述有机胺为乙二胺。
本发明的第四个方面是提供本发明所述胺基富勒烯衍生物在清除自由基中的应用。
所述自由基包括单线态氧自由基、羟基自由基、过氧化氢自由基、超氧阴离子自由基等自由基中的至少之一。
根据本发明,所述应用可以是作为治疗或预防性用药,用于人体或动物体;也可以是非治疗或预防性药物的应用,例如,在某些化学反应条件下,为避免反应体系中出现自由基,产生干扰,在所述反应体系中事先加入所述的胺基富勒烯衍生物。
在本发明的实施例中,在所述胺基富勒烯衍生物清除羟基自由基的研究中,所用胺基富勒烯衍生物的浓度为:20、50、100、200、400μm,浓度在50μm时,就有明显的清除自由基的效果,在400μm时,清除羟基自由的效果可达90%。
本发明的第五个方面是提供所述胺基富勒烯衍生物在制备自由基清除剂中的应用。
所述自由基包括单线态氧自由基、羟基自由基、过氧化氢自由基、超氧阴离子自由基等自由基中的至少之一。
所述自由基清除剂用于保护细胞抗自由基损伤。
在本发明的实施例中,在所述胺基富勒烯衍生物的细胞毒性研究中,所用胺基富勒烯衍生物的浓度在0~400μm范围内,没有细胞毒性,且细胞活性较未加入胺基富勒烯衍生物的对照组的活性高。
在本发明的实施例中,在所述胺基富勒烯衍生物的细胞抗氧化保护实验中,所用胺基富勒烯衍生物的浓度在0~100μm范围内,在2.5μm时就有明显的抗细胞氧化的保护效果。
本发明的优点在于:
1、本发明提供的胺基富勒烯衍生物的制备方法,可以方便地通过调节反应温度或者反应时间,控制获得的胺基富勒烯衍生物中的胺基数量;使后续提纯处理操作简便,仅需采用酸化处理、透析纯化和阴离子交换等,就能够完全除去残留的有机胺和无机杂质,提纯效果好。
2、本发明提供的胺基富勒烯衍生物的制备方法能制备得到水溶性提高的胺基富勒烯衍生物,冷冻干燥后,可以完全重溶于超纯水中;并且粒径小、且表面带正电荷,分散性好、不易发生团聚,适合细胞吞噬,自由基捕获效率高,清除自由基的效果好。
3、与羧酸富勒烯衍生物相比,胺基富勒烯衍生物中的氨基还能够进一步与生物分子如dna、氨基酸发生键合反应,从而可以进一步丰富其在生物医学领域的应用。
4、所制备的胺基富勒烯衍生物在一定的浓度范围内不但没有细胞毒性,且具有很好的细胞保护抗自由基损伤的作用。
附图说明
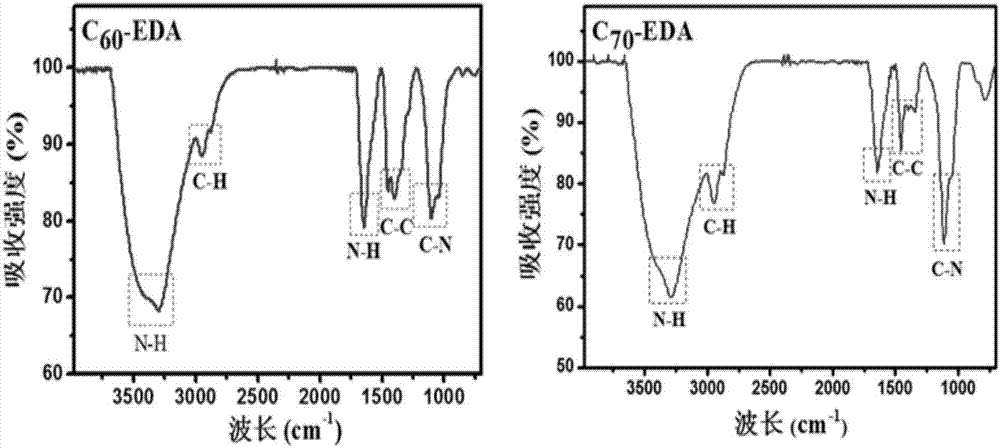
图1显示了本发明实施例1以及实施例2制备的胺基富勒烯衍生物的红外吸收图谱;
图2显示了本发明实施例1以及实施例2制备的胺基富勒烯衍生物的水合粒径和zeta电位;
图3使用esr测试本发明实施例1以及实施例2制备的胺基富勒烯衍生物清除羟基自由基的效果;
图4使用huvec细胞测试本发明实施例1以及实施例2制备的胺基富勒烯衍生物的细胞毒性;
图5使用huvec细胞测试本发明实施例1以及实施例2制备的胺基富勒烯衍生物对保护细胞抗自由基损伤的效果。
具体实施方式
下面将结合实施例对本发明的方案进行解释。本领域技术人员能够理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1:制备c60(eda)6
(a)用分析天平称取50mg固体富勒烯c60(纯度:99%,厦门福纳新材料科技有限公司)溶于25ml的邻二甲苯溶液中,超声分散30min,用量筒量取10ml乙二胺(分析纯,国药试剂),所述邻二甲苯溶液和乙二胺加入250ml具塞锥形瓶再加入磁子,使用磁力搅拌器搅拌1h(温度:室温,转速:1000r/min,n2作保护气),静置、收集沉淀。
(b)在收集到的沉淀中加入50ml的乙二胺(分析纯,国药试剂),搅拌反应24h(温度:室温,转速:1000r/min,n2作保护气)。
(c)将(b)步骤得到的溶液加入250ml的圆底烧瓶中,再使用旋转蒸发仪(型号:ikarv10basic)将反应溶液旋转蒸发干燥完全(温度:60摄氏度,转速:80r/min)。加入超纯水溶解干燥后得到的固体物质,如若有少量的不溶物,加入稀盐酸溶液(浓度为:1mol/l)至圆底烧瓶中,振荡烧瓶使其内壁上的蒸干物溶解在稀盐酸中,得到红棕色澄清溶液。
(d)将步骤(c)得到的溶液使用naoh水溶液(浓度:10mol/l)中和,ph试纸检测为弱酸性(ph为4~6左右),以保证过量的乙二胺以氯化盐形式存在,能够在后续的透析步骤充分除去。将中和后溶液装入透析袋(截止分子量为3500)放入超纯水中透析至超纯水的电导率小于1μs/cm。
(e)将透析后的溶液进行阴离子交换处理,将氯离子完全转化为氢氧根离子,从而将胺基富勒烯盐酸盐转化为胺基富勒烯。
(f)将阴离子交换处理后的溶液再次进行透析至超纯水的电导率小于1μs/cm,即可得到水溶性的胺基富勒烯衍生物。
将红棕色溶液滴于银镜上,自然干燥后用于红外光谱(ir)测试。
如图1所示,样品在3300nm左右的红外特征吸收(-nh2的伸缩振动吸收峰)证明乙二胺被键合到富勒烯的碳笼,同时在800-1500nm红外特征吸收(碳笼上c-c和c=c的伸缩振动峰)的变化也佐证乙二胺已经键合到碳笼上。
将样品溶液直接用于动态光散射(dls)测试,如图2所示,其水合粒径在120nm左右且表面带正电荷,适合被细胞捕获、吞噬。
将样品冷冻干燥用于c、h、n元素分析(ea),随机在样品中选择2个位置,分析结果见表1,由n:c比例可以确定其化学组成为c60(eda)6。
表1:c60(eda)6元素分析结果
通过调整(a)步骤中乙二胺和富勒烯的反应时间,可以调控键合的乙二胺的数量,获得c60(eda)m,m=5~15。
实施例2:制备c70(eda)8
反应条件以及步骤同实施例1,所不同的是,称取50mg富勒烯c70固体(纯度:99%,厦门福纳新材料科技有限公司)替代c60。最终获得含有c70(eda)8的澄清红棕色溶液。
采用与实施例1中相同的测试方法以及条件对获得的产品进行成分检测。
如图1所示,样品在3300nm左右的红外特征吸收(-nh2的伸缩振动吸收峰)证明乙二胺被键合到富勒烯的碳笼,同时在800~1500nm红外特征吸收(碳笼上c-c和c=c的伸缩振动峰)的变化也佐证乙二胺已经键合到碳笼上。
将样品溶液直接用于动态光散射(dls)测试,如图2所示,其水合粒径在140nm左右且表面带正电荷,适合被细胞捕获、吞噬。
将样品冷冻干燥用于c、h、n元素分析(ea),随机在样品中选择2个位置,分析结果见表2,由n:c比例可以确定其化学组成为c70(eda)8。
表2:c70(eda)8元素分析结果
通过调整乙二胺和富勒烯的反应时间,可以调控键合的乙二胺的数量,获得c70(eda)m,m=5~15。
通过富勒烯上的c的含量可以对样品定量,且用超纯水将所制备的c60(eda)6和c70(eda)8配制成不同浓度的水溶性胺基富勒烯衍生物溶液,用于以下实施例中。
实施例3:c60(eda)6、c70(eda)8对羟自由基的清除测试
利用紫外(uv)诱导h2o2产生羟基自由基,以dmpo为自由基捕获剂,dmpo很快与游离羟基自由基结合生成dmpo-oh,可以通过epr检测到。
溶液配制:100μl的dmpo(100mm)与50μl的h2o2(100mm)以及50μl的超纯水混合制得空白对照组。
测试c60(eda)6、c70(eda)8清除自由基时,在测试体系内加入的样品浓度分别为:20、50、100、200、400μm。
测试结果见图3。c60(eda)6、c70(eda)8浓度为50μm时,就有明显的清除自由基的效果,当c60(eda)6、c70(eda)8分别为400μm时,清除羟基自由的效果可达90%,有明显的量效关系。为图示方便,附图3中只呈现了50μm和400μm的结果。
实施例4:细胞毒性测试
使用人脐静脉内皮细胞(huvec)测试实施例1、2中制备的c60(eda)6、c70(eda)8的细胞毒性:
使用含15%小牛血清的dmem培养基对huvec细胞(中国微生物菌种网)进行孵育传代,使用胰蛋白酶消化细胞后,以5×104/ml细胞密度种96孔板(以4孔为1个实验组,第一组为对照组),每孔加200μl细胞分散液,周围一圈的孔中加入磷酸盐缓冲溶液(pbs)在孵箱中孵育24小时;更换为180μl新鲜培养基,分别加入20μl浓度分别为100、200、400μmol/l的c60(eda)6溶液或c70(eda)8溶液,在避光条件下孵育3小时;更换为90μl无色的培养基和10μl的cck-8(细胞活性检测剂),孵育120min至颜色变成亮橙色时,用酶标仪检测细胞活性。
细胞活性结果见图4,加入c60(eda)6或c70(eda)8的实验组细胞活性高于对照组,并且呈现量效关系,即,随着加入c60(eda)6或c70(eda)8的浓度增加,细胞活性增高。由此可见,c60(eda)6、c70(eda)8样品没有表现出任何细胞毒性,而且较对照组细胞活度还有所提高,说明c60(eda)6、c70(eda)8具有很好的细胞保护作用。
实施例5:细胞抗氧化实验
使用人脐静脉内皮细胞(huvec)测试实施例1、2中制备的c60(eda)6、c70(eda)8的细胞抗氧化实验:
使用含15%小牛血清的dmem培养基对huvec细胞(中国微生物菌种网)进行孵育传代,使用胰蛋白酶消化细胞后,以5×104/ml细胞密度种96孔板(以8孔为1个实验组,第一组为对照组,第二组为h2o2对照组),每孔加200μl细胞分散液,周围一圈的孔中加入磷酸盐缓冲溶液(pbs)在孵箱中孵育24小时;更换为90μl新鲜培养基,分别加入10μl浓度分别为2.5、5、10、20、40、80μmol/l的c60(eda)6、c70(eda)8溶液,在避光条件下孵育3小时;更换为100μl的双氧水(3mm)孵育1h,再更换为200μl的细胞培养液孵育24h,最后更换为90μl无色的培养基和10μl的cck-8(细胞活性检测剂),孵育120min至颜色变成亮橙色时,用酶标仪检测细胞活性。
细胞活性结果见图5,采用h2o2处理后细胞的活性均有降低,用c60(eda)6、c70(eda)8做过预处理的细胞样品,其细胞活性降低的程度有所抑制,说明c60(eda)6、c70(eda)8对过氧化氢损伤细胞有一定的保护作用。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
- 还没有人留言评论。精彩留言会获得点赞!