一种艾沙康唑晶型及其制备方法与流程
本发明属于药物化学
技术领域:
,具体而言,涉及一种艾沙康唑的晶型i及其制备方法。
背景技术:
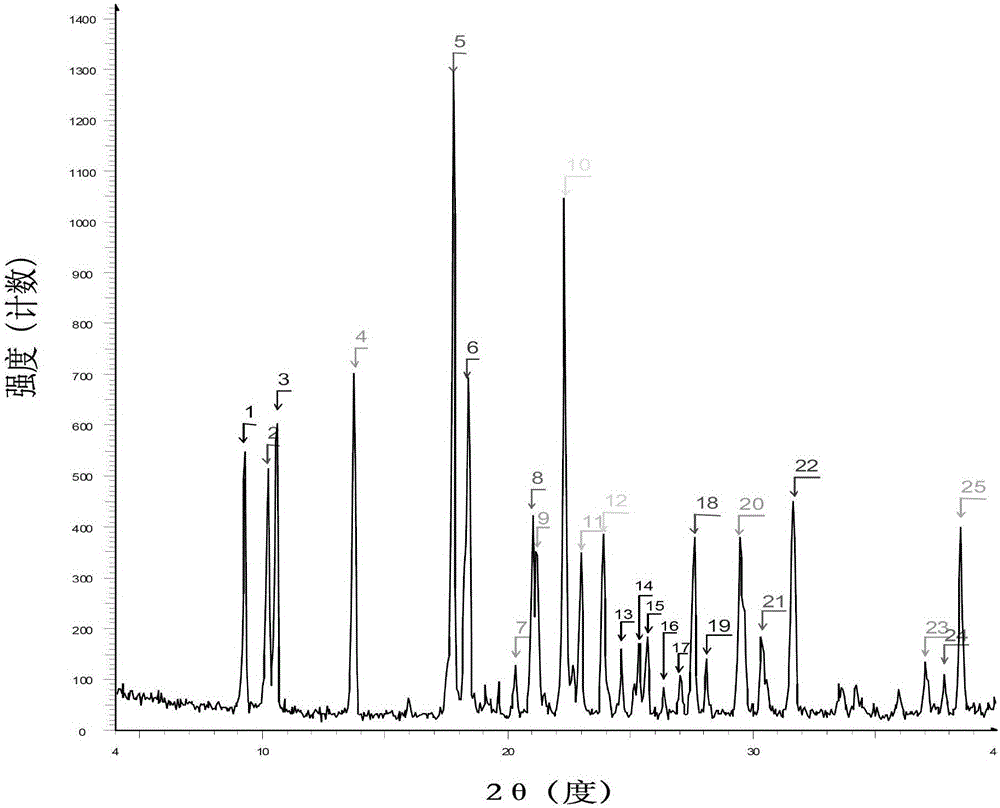
:艾沙康唑(isavuconazole)由安斯泰来与巴塞利亚(basilea)制药公司共同开发,是第三代的三唑类抗真菌药物。每天一次静注或口服的广谱抗真菌制剂,用于侵袭性曲霉菌、毛霉菌、念珠菌等危及生命的真菌感染。通常发生在免疫力低下的病人身上,如化疗中的肿瘤患者、接受免疫抑制治疗的器官移植患者等。其中艾沙康唑硫酸酯(isavuconazoniumsulfate)列入美国、欧盟侵袭性曲霉菌、毛霉菌病的孤儿药(orphanmedicinalproducts),fda于2015年3月6日批准其上市。艾沙康唑硫酸酯(isavuconazoniumsulfate)为艾沙康唑(isavuconazole)的前药。艾沙康唑硫酸酯经静脉注射或口服给药后,可在酯酶的作用下迅速完全转化为艾沙康唑及少量的降解产物。wo2001/032652首次公开了艾沙康唑的结构式、制备方法及用途,其英文名:isavuconazole,结构式如下:该专利以(r)-乳酸甲酯为起始原料,经过11步反应得到艾沙康唑,总收率在16%不到。该路线起始原料包含一个手性中心,通过corey或sharpless环氧方法不对称合成构建第二个手性中心,然而选择性并不够好,得到的异构体也难以纯化,收率较低。wo2003/002498也公开了如下的艾沙康唑的具体制备方法:该专利以(r)-3-丁炔-2-醇为起始物料,在pd(ii)催化剂和二乙基锌的作用下一步合成第二个手性中心,该方法基本能得到ee值80%左右,但起始物料和pd(ii)催化剂昂贵,生产成本大大增加。us2182025公开了新的合成路线,该专利以1-(2,5-二氟苯基)-2-(1h-1,2,4-三唑-1-基)乙酮为起始原料,该原料易得,但第一步需在-78℃下反应,反应较为苛刻,且选择性不高,较难纯化。因此,现有技术中缺乏安全、经济、可大规模且有效(高收率、高纯度)的用于制备艾沙康唑的方法。同时需要开发一种理化性质较优的新晶型,来提高艾沙康唑的稳定性。技术实现要素:本发明的目的之一在于提供一种艾沙康唑的新晶型,将其命名为晶型i,用于治疗侵袭性曲霉菌、毛霉菌、念珠菌等危及生命的真菌感染。对比无定形的艾沙康唑,晶型i的艾沙康唑具有更良好的稳定性。本发明的另一个目的是提供一种艾沙康唑的晶型i的制备方法,所述方法工艺简单,条件温和,大大提高了生产效率,适合工业化生产高纯度的艾沙康唑的晶型i。一方面,本发明提供的艾沙康唑的晶型i,所述晶型i在以2θ角度表示的x-射线粉末衍射图谱(xrpd图谱)在约9.207°±0.2。、10.177°±0.2。、10.507°±0.2。、13.664°±0.2。、17.761±0.2。、18.324°±0.2。、21.063°±0.2。、22.290°±0.2。、31.673°±0.2。、38.538°±0.2。处有特征峰,特别地,所述特征峰相对强度大于30%。特别地,所述的艾沙康唑的晶型i的xrpd图谱,还在衍射角度2θ角约为21.173°±0.2。、22.961°±0.2。、23.888°±0.2。、24.608°±0.2。、25.374°±0.2。、25.662°±0.2。、27.598°±0.2。、28.110°±0.2。、29.518°±0.2。、30.388°±0.2。、37.104°±0.2。处有特征峰,特别地,所述特征峰相对强度大于10%。进一步地,所述的艾沙康唑的晶型i的xrpd图谱,还在衍射角度2θ角约为20.286°±0.2。、26.365°±0.2。、27.080°±0.2。、37.877°±0.2。处有特征峰,特别地,所述特征峰相对强度大于5%。特别地,本发明所述的艾沙康唑的晶型i,具有基本上如附图1所示的xrpd图谱,其中,所述晶型i的xrpd数据见表1。表1序号2θ°相对强度序号2θ°相对强度19.20742.11425.37413210.17739.51525.66214310.50746.51626.3656.2413.664541727.088517.7611001827.58929.2618.32453.41928.1110.5720.2869.62029.51829821.06332.32130.38813.8921.17327.12231.67334.61022.2980.82337.10410.11122.96126.72437.8778.31223.88829.62538.53830.61324.60812.1由于测量条件的不同,xrpd衍射图上各峰2θ角和相对强度会有所变动,一般2θ角变化在±0.2°以内,但也能稍溢出该范围,本领域技术人员应理解,衍射的相对强度可取决于,例如,样品制剂或所用设备。所述的艾沙康唑的晶型i的熔点约为121.97±1℃,特别地,所述艾沙康唑的晶型i具有基本如如图2所示的差示扫描量热分析(dsc)图谱。本发明提供的艾沙康唑的晶型i具有良好的熔点和质量,其质量纯度至少为99.0%,ee值至少为99.0%。艾沙康唑的晶型i具有大规模合成或配制成治疗用制剂所需的性能,对光、湿、热稳定,便于生产、储存。另一方面,本发明提供了一种制备高纯度艾沙康唑的晶型i的方法,所述制备方法包括:(d)将艾沙康唑粗产品加入强极性溶剂中加热溶解,得溶液;(e)向步骤(d)制得的溶液中,加入不良溶剂,自然降温至析出晶体的温度析出晶体,过滤,干燥得艾沙康唑的晶型i;步骤(d)中,所述的艾沙康唑粗产品可以是任何工艺得到的艾沙康唑粗产品,包括艾沙康唑的无水形式、溶剂合物或水合物形式的各种已知或未知的晶型或无定型形式的工业粗产品;所述的强极性溶剂为选自甲醇、乙醇、异丙醇、四氢呋喃中的一种或几种,优选地,所述的强极性溶剂为乙醇;所述艾沙康唑粗产品与强极性溶剂的重量体积比为1kg:(2~3)l;所述的溶解的温度为40~80℃,优选地,所述的溶解的温度为60~65℃;步骤(e)中,所述不良溶剂为选自石油醚、正己烷、甲基叔丁基醚、水中的一种或几种,优选地,所述的不良溶剂为水;所述艾沙康唑粗产品与不良溶剂的重量体积比为1kg:(1~3)l;所述析出晶体的温度为0~25℃,优选地,所述的析出晶体的温度为10~25℃。发明人在艾沙康唑的研究中发现,升高温度可大大减小步骤(d)中艾沙康唑所需的溶剂体积,从而降低生产成本,并且步骤(e)的析出晶体过程降温有利于迅速析出晶体,使生产效率提高。此外,为了解决艾沙康唑的晶型i制备方法中有色杂质含量过多,有关物质超标,并影响析出晶体的问题,本发明的制备方法步骤(d)中还加入少量活性炭进行脱色,优选地,所述艾沙康唑粗产品与活性炭的重量比为1:0.05。经脱色后得到的晶型产品,色泽纯净,外观均一。为进一步提高工业生产效率、减少溶剂用量,降低生产成本,发明人对本发明制备方法条件进行了优化。优选地,所述艾沙康唑的晶型i的制备方法,在步骤(d)之前还包括如下步骤:(a)在溶剂中,将硫代酰胺ⅱ和2-溴-4'-氰基苯乙酮加入,升温发生缩合反应,得反应液;(b)浓缩步骤(a)的反应液,在有机溶剂中结晶,过滤,得艾沙康唑氢溴酸盐;(c)将步骤(b)制得的艾沙康唑氢溴酸盐用碱处理,萃取,得到艾沙康唑粗产品。步骤(a)中,所述缩合反应的反应温度为30~65℃,优选为40~45℃,反应时间一般为20~24小时;用hplc检测反应终点;所述的溶剂为选自石油醚、正己烷、甲基叔丁基醚、乙醇中的一种或几种,优选为乙醇;所述硫代酰胺ⅱ与2-溴-4'-氰基苯乙酮的摩尔比为1:0.95~1:1.3,优选为1:1.05~1:1.1。步骤(b)中,所述有机溶剂为选自石油醚、正己烷、甲基叔丁基醚、乙酸乙酯中的一种或几种,优选为乙酸乙酯;所述结晶为两段结晶,先在40~55℃析出晶体,然后再降温至15~25℃继续析出晶体;所述结晶的时间为3~24小时;优选地,所述的结晶为在40~55℃,搅拌析出晶体1~4h,自然降至15~25℃,继续析出晶体2~6h;步骤(c)中,所述的碱为选自碳酸氢钠、碳酸钠、氢氧化钠中的一种或几种,优选为碳酸氢钠、碳酸钠。有益效果由于以上技术方案的实施,本发明与现有技术相比具有如下优点:本发明提供了一种新的艾沙康唑晶型i,为艾沙康唑在医药工业的应用提供了新的选择,本发明还提供一种制备艾沙康唑晶型i的方法,该方法操作简便,条件温和,所用试剂廉价易得,适于工业化生存,所得晶型i纯度高,收率高,杂质含量、ee值符合药用要求。附图说明图1是实施例6中制备的艾沙康唑晶型i的x-射线粉末衍射xrpd图谱;图2是实施例6中制备的艾沙康唑晶型i的热重分析tga图谱;图3是实施例6中制备的艾沙康唑晶型i的差示扫描量热分析dsc图谱;图4是对比例中制备的艾沙康唑无定型物的x-射线粉末衍射xrpd图谱。具体实施方式本发明进一步参考以下实施例,所述实施例详细描述本发明的盐及晶型的制备和应用。对本领域技术人员显而易见的是,对于材料和方法两者其中的许多改变可在不脱离本发明范围的情况下实施。检测仪器及方法:x-射线粉末衍射(xrpd)所使用的仪器为brukerd8advancediffractometer,采用铜靶波长为1.54nm的kαx-射线,在40kv和40ma的操作条件下、θ-2θ测角仪、mo单色仪、lynxeye探测器。仪器在使用前用仪器自带的标准样品校正峰位。采集软件是diffracplusxrdcommander,分析软件是mdijade5.0。样品在室温条件下测试,把需要检测的样品放在有机玻片上。详细检测条件如下:角度范围:3~40°2θ;步长:0.02°2θ;速度:0.2秒/步。除非特别说明,样品在检测前未经研磨。热重分析(tga)数据采自于tainstrumentsq500tga,仪器控制软件是thermaladvantage,分析软件是universalanalysis。通常取5~15mg的样品放置于白金坩埚内,采用分段高分辨检测的方式,以10℃/min的升温速度在50ml/min干燥n2的保护下将样品从室温升至300℃,同时ta软件记录样品在升温过程中的重量变化。差热分析(dsc)数据采自于tainstrumentsq200mdsc,仪器控制软件是thermaladvantage,分析软件是universalanalysis。通常取1~10毫克的样品放置于加盖打孔(除非特别说明)的铝坩埚内,以10℃/min的升温速度在50ml/min干燥n2的保护下将样品从室温升至200℃或300℃,同时ta软件记录样品在升温过程中的热量变化。在本申请中,熔点是按起始温度来报告的。所有温度以℃(摄氏度)表示,室温是指20~25℃。2-溴-4’-氰基苯乙酮,乙醇,乙酸乙酯等试剂均为分析纯,由国药集团化学试剂有限公司提供,所用试剂和溶剂除特别说明外,均未经特别处理。硫代酰胺ⅱ参考cn1223564c中实施例14的制备方法。艾沙康唑购自上海皓元医药股份有限公司,纯度:98.9%。hplc纯度测定条件仪器:高效液相色谱仪配备紫外检测器色谱柱:watersxbridgec18150×4.6mm,3.5μm流动相a:0.01mol/l磷酸二氢钾溶液(ph2.8):乙腈=100:10,v:v流动相b:乙腈检测波长:266nm/210nm流速:0.7ml/min进样量:10μl柱温:40℃运行时间:40min梯度表:稀释液:乙腈:水=50:50,v:v供试品溶液:精密称取供试品适量,用稀释液溶解并稀释成每1ml中约含0.5mg的溶液。对照溶液:精密量取供试品溶液适量,用稀释液稀释成每1ml中约含5ug的溶液程序/系统适用性要求:按照色谱条件分别进样空白溶液、对照溶液和供试品溶液,记录色谱过程。要求空白溶液无干扰,重复进6针对照溶液,在6针对照溶液所得图谱中,主峰峰面积计的rsd≤2.0%。杂质含量计算:用主成分自身对照法计算样品中杂质含量hplc手性测定条件:仪器:高效液相色谱仪配备紫外检测器色谱柱:chiralpakic250×4.6mm,5μm流动相:0.1%正丁胺[正己烷:乙醇:甲醇=20:4:1,v:v:v]检测波长:266nm流速:1.0ml/min进样量:20μl柱温:30℃运行时间:40min稀释液:异丙醇供试品溶液:精密称取供试品适量,用稀释液溶解并稀释成每1ml中约含1.0mg的溶液。对照溶液:精密量取供试品溶液适量,用稀释液稀释成每1ml中约含10μg的溶液。分离度溶液:精密称取艾沙康唑原料药,艾沙康唑对映异构体3c(4-[2-[(2s,3s)-3-(2,5-二氟苯基)-3-羟基-4-(1,2,4-三唑-1-基]-1,3-噻唑-4-基]苯甲腈)和非对映异构体3b(4-[2-[(2r,3s)-3-(2,5-二氟苯基)-3-羟基-4-(1,2,4-三唑-1-基]-1,3-噻唑-4-基]苯甲腈)、3d(4-[2-[(2s,3r)-3-(2,5-二氟苯基)-3-羟基-4-(1,2,4-三唑-1-基]-1,3-噻唑-4-基]苯甲腈)标准品(以上三个异构体标准品均购于深圳市恒丰万达生物科技有限公司)适量,用稀释液溶解并制成每1ml中约含各组分均为10μg的混合溶液。程序/系统适用性要求:按照色谱条件依次分别进样空白溶液、分离度溶液、对照溶液和供试品溶液,记录色谱过程。要求空白溶液无干扰,分离度溶液各组分分离度不得小于3.0,重复进样5针对照溶液,在5针对照溶液所得图谱中,主峰峰面积计的rsd≤2.0%。杂质计算:以1%自身对照法计算异构体含量实施例1艾沙康唑氢溴酸盐的制备于500ml反应瓶中加入190ml无水乙醇和19.0g硫代酰胺ii,升温至40~45℃,搅拌溶清后,加入12.95g2-溴-4’-氰基苯乙酮,在此温度下反应20~24h。在40~50℃下减压浓缩至干,加入190ml乙酸乙酯后升至50~55℃,搅拌析出晶体2h后,自然降至15~25℃,继续搅拌析出晶体4h。过滤,干燥得21.1g艾沙康唑氢溴酸盐,收率:67.1%,hplc:98.72%。1hnmr(400mhz,dmso-d6)δ:8.74(s,1h),8.46(s,1h),8.22(d,j=8.4hz,2h),8.01(s,1h),7.94(d,j=8.4hz,2h),7.63(br,2h),7.26-7.33(m,1h),7.17-7.22(m,1h),7.04-7.09(m,1h),4.96(d,j=14.48hz,1h),4.51(d,j=14.48hz,1h),4.15(dd,j1=14.48hz,j2=7.12hz,1h),1.19(d,j=7.12hz,3h).实施例2艾沙康唑氢溴酸盐的制备于500ml反应瓶中加入190ml无水乙醇和19.0g硫代酰胺ii,升温至60~65℃,搅拌溶清后,加入14.99g2-溴-4’-氰基苯乙酮,在此温度下反应20~24h。在40~50℃下减压浓缩至干,加入190ml乙酸乙酯后升至50~55℃,搅拌析出晶体2h后,自然降至15~25℃,继续搅拌析出晶体4h。过滤,干燥得19.0g艾沙康唑氢溴酸盐,收率:60.5%,hplc:97.37%。实施例3艾沙康唑氢溴酸盐的制备于500ml反应瓶中加入190ml无水乙醇和19.0g硫代酰胺ii,升温至40~45℃,搅拌溶清后,加入14.25g2-溴-4’-氰基苯乙酮,在此温度下反应20~24h。在40~50℃下减压浓缩至干,加入190ml乙酸乙酯后升至50~55℃,搅拌析出晶体2h后,自然降至15~25℃,继续搅拌析出晶体4h。过滤,干燥得22.0g艾沙康唑氢溴酸盐,收率:69.8%,hplc:98.07%。实施例4艾沙康唑的晶型i的制备于500ml反应瓶中加入220ml乙酸乙酯和实施例1制备的21.0g艾沙康唑氢溴酸盐,用210ml饱和碳酸氢钠中和、分层,有机相用210ml饱和食盐水洗涤,无水硫酸钠干燥。过滤,滤液在40~50℃下减压浓缩至干,加入42ml无水乙醇,升温至60~65℃搅拌溶清后加入1.05g活性炭,保温1h,热过滤,滤饼用10ml无水乙醇洗涤。将滤液升温至60~65℃,在此温度下缓慢滴加26ml纯化水,继续保温1h,然后自然降至15~25℃继续搅拌析出晶体24h,过滤,滤饼用21ml乙醇:水(2:1,v/v)洗涤,得到的滤饼在40~50℃减压干燥8h,得14.0g类白色固体,xrpd确认为艾沙康唑晶型i。收率:75.4%,hplc纯度:99.89%,ee值:99.92%。1hnmr(400mhz,dmso-d6)δ:8.41(s,1h),8.25(s,1h),8.20(d,j=8.52hz,2h),7.92(d,j=8.52hz,2h),7.65(s,1h),7.22-7.28(m,1h),7.13-7.18(m,1h),7.04-7.09(m,1h),6.15(s,1h),4.92(d,j=14.48hz,1h),4.41(d,j=14.48hz,1h),4.14(dd,j1=14.48hz,j2=7.2hz,1h),1.17(d,j=7.2hz,3h).实施例5艾沙康唑的晶型i的制备于500ml反应瓶中加入190ml乙酸乙酯和实施例2制备的19.0g艾沙康唑氢溴酸盐,用190ml饱和碳酸氢钠中和、分层,有机相用190ml饱和食盐水洗涤,无水硫酸钠干燥。过滤,滤液在40~50℃下减压浓缩至干,加入38ml无水乙醇,升至60~65℃搅拌溶清后加入0.95g活性炭,保温1h,热过滤,滤饼用9ml无水乙醇洗涤。将滤液升温至60~65℃,在此温度下缓慢滴加47ml纯化水,继续保温1h,然后自然降至10~15℃继续搅拌析出晶体24h,过滤,滤饼用19ml乙醇:水(1:1,v/v)洗涤,得到的滤饼在40~50℃减压干燥8h,得14.85g类白色固体,xrpd确认为艾沙康唑晶型i。收率:92.6%,hplc纯度:99.80%,ee值:99.90%。实施例6艾沙康唑的晶型i的制备于500ml反应瓶中加入220ml乙酸乙酯和实施例3制备的22.0g艾沙康唑氢溴酸盐,用220ml饱和碳酸氢钠中和、分层,有机相用220ml饱和食盐水洗涤,无水硫酸钠干燥。过滤,滤液在40~50℃下减压浓缩至干,加入44ml无水乙醇,升温至60~65℃搅拌溶清后加入1.1g活性炭,保温1h,热过滤,滤饼用11ml无水乙醇洗涤。将滤液升温至60~65℃,在此温度下缓慢滴加55ml纯化水,继续保温1h,然后自然降至15~25℃继续搅拌析出晶体24h,过滤,滤饼用22ml乙醇:水(1:1,v/v)洗涤,得到的滤饼在40~50℃减压干燥8h,得17.0g类白色固体,即艾沙康唑晶型i。收率:91.6%,hplc纯度:99.95%,ee值:99.91%。测定其的xrpd图谱、tga图谱及dsc图谱,分别见图1、图2和图3,其与实施例4和5中的艾沙康唑晶型i的xrpd图谱、tga图谱及dsc图谱基本一致,由此可见,其确为艾沙康唑晶型i。其中,图1的xrpd图谱的具体数据如表2所示。表2实施例7于500ml反应瓶中加入190ml无水乙醇和19.0g硫代酰胺ii,升温至40~45℃,搅拌溶清后,加入14.25g2-溴-4’-氰基苯乙酮,在此温度下反应20~24h。在40~50℃下减压浓缩至干,加入190ml乙酸乙酯后升至50~55℃,搅拌析出晶体2h后,自然降至室温,继续搅拌析出晶体4h。过滤,得到的滤饼加入至有220ml乙酸乙酯的反应瓶中,用220ml饱和碳酸氢钠中和、分层,有机相用220ml饱和食盐水洗涤,无水硫酸钠干燥。过滤,滤液在40~50℃下减压浓缩至干,加入44ml无水乙醇,升温至60~65℃搅拌溶清后加入1.1g活性炭,保温1h,热过滤,滤饼用11ml无水乙醇洗涤。将滤液升温至60~65℃,在此温度下缓慢滴加55ml纯化水,继续保温1h,然后自然降至15~25℃继续搅拌析出晶体24h,过滤,滤饼用22ml乙醇:水(1:1,v/v)洗涤,得到的滤饼在40~50℃减压干燥8h,得17.2g类白色固体,即艾沙康唑晶型i。收率:61.9%,hplc纯度:99.96%,ee值:99.93%。实施例8于500ml反应瓶中加入20.0g购自上海皓元医药股份有限公司的艾沙康唑粗品和60ml无水乙醇,升温至60~65℃搅拌溶清后,在此温度下缓慢滴加60ml纯化水,继续保温1h,然后自然降至15~25℃继续搅拌析出晶体24h,过滤,滤饼用22ml乙醇:水(1:1,v/v)洗涤,得到的滤饼在40~50℃减压干燥8h,得18.2g类白色固体,即艾沙康唑晶型i。收率:91.0%,hplc纯度:99.96%,ee值:99.98%。对比例取实施例6制备的10g艾沙康唑晶型i,将其溶于100ml二氯甲烷溶液中,在35~40℃下减压浓缩至干,得艾沙康唑的无定型物。测定其的xrpd图谱,见图4。测试例另外,为了更好地说明本发明的艾沙康唑晶型i具有良好的稳定性,对实施例4制备的艾沙康唑晶型i和对比例自制的艾沙康唑的无定型物进行了对比实验。稳定性测试方法:引湿性:按照中国药典2015年版四部9103药物引湿性试验指导原则进行试验;高湿试验:取样品适量置于称量瓶中,开口放置于25℃/90%±5%rh条件下,检测第5天和10天的纯度、手性及含量,与第0天样品比较;高温试验:取样品适量置于称量瓶中,开口放置于60℃恒温箱中,检测第5天和10天的纯度、手性及含量,与第0天样品比较;光照试验:取样品适量置于称量瓶中,开口放置于照度4500lx±500lx的光照箱中,检测第5天和10天的纯度、手性及含量,与第0天样品比较。加速试验:取样品适量置于称量瓶中,开口放置于40±2℃/75%±5%rh条件下,检测1m、2m和3m的纯度、手性及含量。通过试验对比艾沙康唑无定型物与本发明的艾沙康唑晶型i,得到的结果如下:表3表4由上表3和表4可见,本发明的艾沙康唑晶型i在高温、高湿、光照条件下稳定性较艾沙康唑无定型更好,且在3m的加速试验中也具有更良好的稳定性。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1