壬二胺的合成方法与流程
本发明属于化学合成领域,具体而言,涉及一种以1,5-戊二醛为原料合成壬二胺的合成方法。
背景技术:
壬二胺又称作1,9-二氨基壬烷、1,9-壬烷二胺、伸壬基二胺,英文名称1,9-nonanediamine、1,9-diaminononane、1,9-nonamethylene-diamine。
壬二胺的主要是作为pa9t的原料。pa9t数值是新型耐热性聚酰胺树脂,目前这种耐热性树脂正处于市场化发展阶段,只有少数厂商能实现工业化,国内无工业化报道。pa9t具有优良的低吸水性、高刚性、耐药用溶剂型、韧性、耐热性、尺寸稳定性和优良的成形性等诸多优点。相比常见的高性能塑料,例如聚苯硫醚(pps)、pa46、pa6t等具有一些其他的性能优势。
同时,壬二胺还可以作为精细化学品的合成原料及聚肽胺、聚氨蜡的合成原料,也可以作为物质改性原料,因此其市场规模日趋扩大。
目前,壬二胺的合成方法主要包括以下几种:
一、日本可乐丽公司:
目前,日本可乐丽株式会社提出了一种合成壬二胺的方法,其合成工艺以丁二烯为原料,在专利us4417079中公开了1,3-丁二烯经过催化剂水合、加氢还原制备2,7-二烯-1-辛醇;在专利us4510331公开了所述醇经过亚铬酸铜的催化剂的催化下氧化生成7-烯-1-辛醛,以及在专利ep1489087中公开了所述醛在高压条件下用一氧化碳羰基化生成壬二醛,在专利jp58167547中公开了1,9-壬二醛和2-甲基-1,8-辛二醛的混合物在高压催化下还原胺化生成壬二胺和2-甲基辛二胺的混合物,经过分离制备壬二胺。反应的化学方程式如下:
然而该工艺存在以下缺点:粗产品中的壬二胺含量低,并且含有异构体;以石油基产品为原料限制了产品在医药及食品领域的应用。
二、叠氮化工艺:
《化学研究与应用》:2003,15(6)p865-867介绍了辛二胺的重氮化合成工艺,使用辛二酸与叠氮化钠在催化剂的作用下生成羰基叠氮化物,再用碱调节ph值至12生成辛二胺,收率约75%左右。但该工艺使用叠氮化钠具有较高的危险性,不适宜大规模工业生产。
三、壬二酸工艺:
中国发明专利公开cn012701991a公开了一种以壬二酸为原料合成壬二胺的合成工艺。
首先以壬二酸为原料在二氯亚砜的作用下生成相应的酰氯,酰氯与氨水生成相应的二酰胺,再经过脱水、加氢还原制备壬二胺。
该工艺用反应方程式表示如下:
该工艺大量使用二氯亚砜,会产生大量的二氧化硫和氯化氢气体,污染严重,同时会对设备造成较大的腐蚀。采用加氢工艺也具有一定的安全隐患。该工艺步骤繁琐,污染严重且具有一定的安全风险,不适宜进行工业化。
技术实现要素:
针对国内尚不能实现壬二胺的工业化生产,文献报道的生产工艺存在“三废”产生量大,壬二胺含量低,以及不能应用于食品领域,对生产设备要求高的问题,经过深入广泛的研究,本发明人提出了壬二胺的新合成工艺。
因此,本发明的目的是提供一种壬二胺的合成方法。本发明的壬二胺合成方法能够解决目前技术面临的“三废”产生量大的难题,同时得到高纯度的壬二胺,扩大其应用范围,同时降低了对生产设备的要求。该方法具有操作简便、生产成本低等诸多优点,能打破国外技术垄断实现国内自给自足。
根据本发明的一个方面,提供一种壬二胺的合成方法,如下面的反应方程式所示:
其中,r彼此相同或不同,各自独立地为h、c1~c20烷基、3~8元环烷基、3~8元环烷基c1~c10烷基、3~8元杂环基、3~8元杂环基c1~c10烷基、5~8元芳基、5~8元芳基c1~c10烷基、5~8元杂芳基或5~8元杂芳基c1~c10烷基;优选为h、c1~c10烷基、3~8元环烷基、3~8元环烷基c1~c8烷基、5~6元杂环基、5~6元杂环基c1~c8烷基、5~6元芳基、5~6元芳基c1~c8烷基、5~8元杂芳基或5~8元杂芳基c1~c8烷基;更优选为h、c1~c6烷基、3~6元环烷基、3~6元环烷基c1~c6烷基、5~6元杂环基、5~6元杂环基c1~c6烷基、5~6元芳基、5~6元芳基c1~c6烷基、5~8元杂芳基或5~8元杂芳基c1~c6烷基;最优选为h、甲基、乙基、丙基、异丙基、正丁基、异丁基、叔丁基、苯基、苄基或环丙基甲基;
1)在步骤1中,在酸或碱催化剂的存在下,1,5-戊二醛与氰乙酸或氰乙酸酯通过缩合反应生成中间体a;
2)在步骤2中,在无催化剂的存在下或在无机碱或有机胺的催化剂的存在下,中间体a通过直接进行脱羧反应或先水解后脱羧反应得到中间体b,中间体b可以是其中的一种或几种异构体的混合物;
3)在步骤3中,在氢化还原催化剂的存在下,中间体b通过加氢反应生成最终产品1,9-壬二胺。
在本发明所述方法的步骤1中,与1,5-戊二醛进行缩合反应的原料可以是选自氰乙酸和氰乙酸酯的一种或两种混合物,所述氰乙酸酯优选包括,但不限于,氰乙酸甲酯、氰乙酸乙酯、氰乙酸丙酯、氰乙酸异丙酯、氰乙酸丁酯、氰乙酸异丁酯、氰乙酸叔丁酯、氰乙酸苄酯等。
在本发明所述方法的步骤1)中,所选用的反应溶剂可以是质子性溶剂、极性非质子性溶剂,也可以是非极性溶剂。其中质子性溶剂包括水、醇类、酸类等;极性非质子溶剂包括酰胺类溶剂、亚砜类溶剂、砜类溶剂等;非极性溶剂包括苯类溶剂、卤代烷类溶剂。具体实例包括但不限于水,甲醇、乙醇、异丙醇、叔丁醇等醇类,二甲基亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、环丁砜、n-甲基吡咯烷酮、丙酮、乙腈、乙酸乙酯、乙酸丁酯、乙酸异丙酯、二氯甲烷、1,2-二氯乙烷、甲苯、吡啶、哌啶;优选地溶剂包括n,n-二甲基甲酰胺、乙酸乙酯和甲醇。
在本发明所述方法的步骤1)中,反应温度为0℃-150℃,优选地反应温度为10℃-100℃。
在本发明所述方法的步骤1)中,所选用的催化剂种类较多,所有可以催化脑文格反应的催化剂均可用于该步反应,可以是酸也可以是碱,优选为碱,因为碱通常具有更好的催化效果,碱可以是有机碱也可以是无机碱。有机碱可以包括2级胺、3级胺,具体的实例包括但不限于二甲胺、哌啶、吗啉、吡啶、三乙胺、二乙胺、1,8-二氮杂二环十一碳-7-烯(dbu)、二异丙基乙基胺等;无机碱包括但不限于氢氧化钠、氢氧化锂、氢氧化钾、氢氧化钙、氧化镁、氧化锆、氧化锌或γ-fe2o3;还包括各种碱性树脂及杂多酸类化合物。优选地,基于戊二醛的质量,所述催化剂的用量为0.1当量到2.0当量,即,催化剂与1,5-戊二醛的摩尔比为0.1~2。
本发明所述方法的步骤1)中,与戊二醛进行缩合反应的化合物包括但不限于氰乙酸、氰乙酸甲酯、氰乙酸乙酯、氰乙酸异丙酯、氰乙酸丁酯,戊二醛与以上氰乙酸或氰乙酸酯化合物的摩尔比为1:2至1:5,优选地摩尔比为1:2-1:3,更优选的摩尔比为1:2-1:2.3。
本发明所述方法的步骤1)中,反应时间为20分钟至24小时,优选地反应时间为1小时-5小时,更优选地反应时间为2-3小时。
本发明所述方法的步骤2)中,所使用的催化剂为无机碱或有机胺,优选三级胺。当选用有机胺作为反应的催化剂时,三级胺相比二级胺具有更好的效果。所述无机碱包括但不限于氢氧化钠、氢氧化锂、氢氧化钾、氢氧化钙、氧化镁、氧化锆、氧化锌或γ-fe2o3。所述有机胺包括但不限于,三乙胺、1,8-二氮杂二环十一碳-7-烯(dbu)、二异丙基乙基胺、吡啶和哌啶。步骤2可以在无催化剂的条件下通过升温实现,加入催化剂会加快反应速度。优选地,基于中间体a的质量,催化剂的加入量可以为0.1当量到10当量;更优选地,碱的加入量为1~2.0当量。即,催化剂与中间体a的摩尔比为0.1~10,优选1~2。
在r不为h时,酯水解和脱羧反应可以采用“一锅法”完成。其中酯的水解过程随着碳链的增长所需反应时间会增长,所需反应温度更高。水解-脱羧反应需要的反应时间为30分钟至10小时,更优选的反应时间为1小时至3小时。水解-脱羧反应温度为30℃至100℃,更优选的反应温度为50℃至70℃。本发明所述方法的步骤2中,当r不为h时,水解反应需要加入水,水的加入的量2-10当量,优选的水的加入量为2-3当量,即,加入的水与1,5-戊二醛的摩尔比为2~10,优选为2~3。
本发明所述方法的步骤2)中,当r为h时,
脱羧反应温度为30℃至200℃,更优选的反应温度为50℃至120℃;
脱羧反应时间为30分钟至10小时,更优选的反应时间为1小时至3小时;脱羧反应的ph=1至14,优选的ph=7-14,更优选的ph=8-10。
本发明所述方法的步骤2)中,脱羧反应的溶剂可以是质子性溶剂、极性非质子溶剂也可以是非极性溶剂,可以是单一溶剂也可以是几种溶剂的混合物。具体的应用实例包括但不限于:水、甲醇、乙醇、异丙醇、正丁醇、叔丁醇、二甲基亚砜、n-甲基吡咯烷酮、n,n-二甲基甲酰胺、n,n-二甲基甲苯胺、乙腈、四氢呋喃、甲基四氢呋喃、吡啶、哌啶、三乙胺、1,2-二氯乙烷、1,1-二氯乙烷等。
本发明所述方法的步骤3)中,氢化还原催化剂主要为金属催化剂,具体的应用实例包括但不限于5%pd/c、10%pd/c、兰尼镍(raney-ni),pt/c等;
本发明所述方法的步骤3)中,催化剂的加入量质量分数(相对于中间体b)0.1‰-30%,优选地质量分数为1‰-10%,更优选的质量分数为5‰-5%;
本发明所述方法的步骤3)中,还原反应在极性非质子性溶剂、质子性溶剂和非极性溶剂中均能进行,但溶剂不同反应速度不同。其中质子性溶剂包括水、醇类、酸类等;极性非质子溶剂包括酰胺类溶剂、亚砜类溶剂、砜类溶剂等;非极性溶剂包括苯类溶剂、卤代烷类溶剂。具体的应用实例包括但不限于:水、甲醇、乙醇、异丙醇、正丁醇、甲酸、醋酸、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n,n-二甲基甲苯胺、n-甲基吡咯烷酮、二甲基亚砜、环丁砜、苯、甲苯、二甲苯、二氯甲烷、1,2-二氯乙烷等。优选的溶剂为甲醇、乙醇。
本发明所述方法的步骤3)中,还原反应氢气压力为常压至10mpa,优选的反应压力为1mpa至5mpa,更优选地反应压力为3mpa。
本发明所述方法的步骤3)中,反应温度10℃至100℃,优选地反应温度为30℃-50℃。
本发明所述方法的步骤3)中,反应时间30分钟至12小时,优选地反应时间为1小时至7小时,更优选的反应时间为3小时至4小时。
其中,在本说明书中,烷基包括直链和支链烷基;
杂环基是指在分子中含有选自n、o和s中的至少一个杂原子的非芳香性环基团,其可以任选被选自c1~c10烷基或c1~c10烷氧基中的一个或多个基团取代,其非限定性的示例包括,但不限于,四氢吡咯基、二氢吡啶基、哌啶基、丙基取代的哌啶基或哌啶基乙基等;芳基是指在分子中不含有杂原子的芳香性环基团,其可以任选被选自c1~c10烷基或c1~c10烷氧基中的一个或多个基团取代,其非限定性的示例包括,但不限于,苯基、苄基等;
杂芳基是指在分子中含有选自n、o和s中的至少一个杂原子的芳香性环基团,其可以任选被选自c1~c10烷基或c1~c10烷氧基中的一个多个基团取代,其非限定性的示例包括,但不限于,甲基吡咯基、吡啶基、甲基吡啶基或咪唑基等。
附图说明
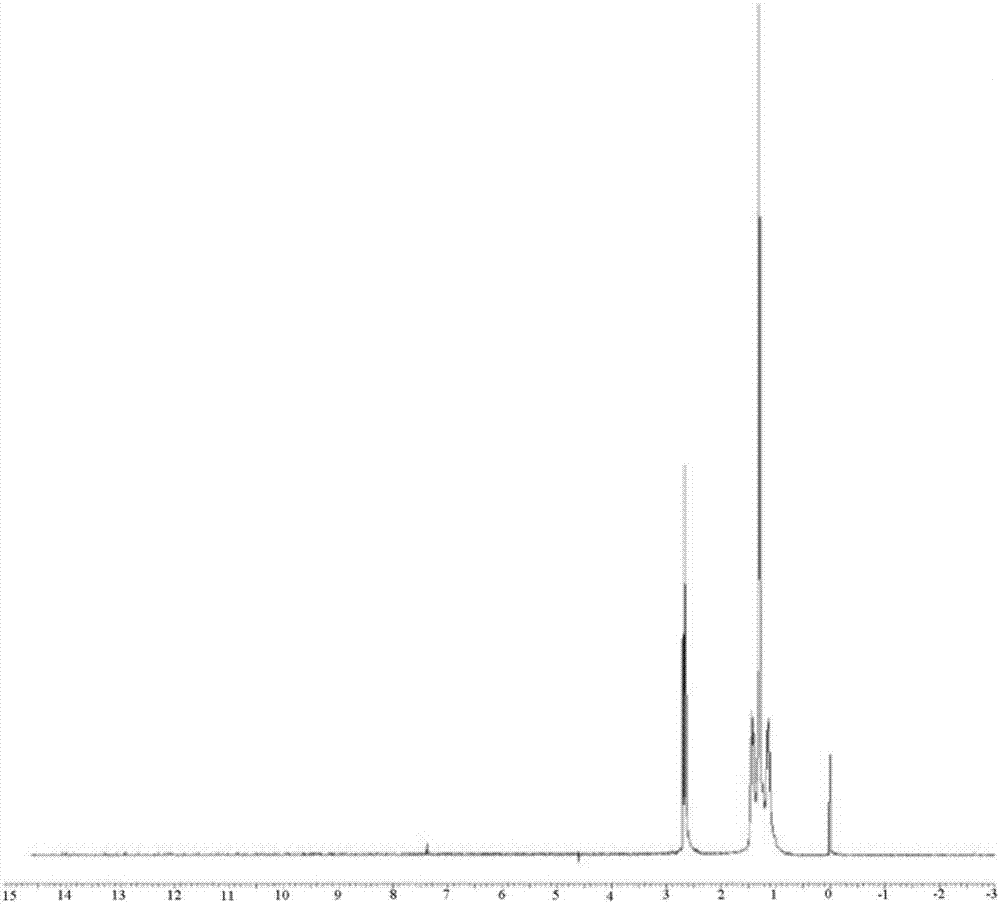
图1为根据本发明的实施例1制备的1,9-壬二胺的1h-nmr图谱。
具体实施方式
实施例1
步骤1:100克戊二醛水溶液(质量分数=50%)用400毫升乙酸乙酯萃取、分液,将乙酸乙酯相倒入2000毫升三口瓶中,冰浴降温至内温低于5℃,称取氰乙酸85克缓慢加入到所述乙酸乙酯溶液中,快速搅拌至完全溶解。称量哌啶21.25克,通过恒压滴液漏斗缓慢加入到反应体系,滴加过程中有热放出,维持体系内温不高于5℃,滴加时间约30分钟。滴加完成后将反应体系自然升温至室温,通过tlc监控反应直至反应完全。通过旋转蒸发的方式蒸干溶剂,得到黄色液体为中间体a粗品,即2,8-二氰基-2,7-二烯-1,9-壬二酸,该产品直接用于下一步反应。
步骤2:向步骤1所得到粗品中加入三乙胺600毫升,加热至回流并持续反应5小时后停止加热,体系分层,待反应体系降温至室温后分离出下层,得到中间体b,该中间体可直接用于下步反应。
步骤3:将步骤2所得中间体b的粗品溶于600毫升甲醇中并转移到加氢反应釜中,向反应体系中分别加入氢氧化钠1.1g,兰尼镍7.3克,封闭反应釜并先后用氮气、氢气各置换三次,最后保持釜内氢气压力3mpa到3.5mpa,反应温度在30分钟内由25℃升温至75℃,搅拌速度为500转/分钟,反应维持4小时直至反应完全。待反应体系将至室温,通过精馏的方式进行产品纯化,最终得到1,9-壬二胺71.5克,总收率为90.5%,其1h-nmr(300mhz,cdcl3)图谱参见图1。
实施例2
步骤1:100克戊二醛水溶液(质量分数=50%)用400毫升乙酸乙酯萃取、分液,将乙酸乙酯相倒入2000毫升三口瓶中,冰浴降温至内温低于5℃,称取氰乙酸甲酯99克缓慢加入到所述乙酸乙酯溶液中。称量三乙胺50.5克,通过恒压滴液漏斗缓慢加入到反应体系,滴加过程中有热放出,维持体系内温不高于5℃,滴加时间约10分钟。滴加完成后将反应体系自然升温至室温,通过tlc监控反应直至反应完全。待反应完全,通过旋转蒸发的方式蒸干溶剂,得到黄色液体为中间体a粗品,即2,8-二氰基-2,7-二烯-1,9-壬二酸二甲酯,该产品直接用于下一步反应。
步骤2:向步骤1所得到粗品中加入三乙胺300毫升、乙腈300ml、水27毫升,加热至回流并持续反应7小时后停止加热,待体系温度降至室温,减压旋蒸蒸干溶剂,得到中间体b的粗品,该中间体可直接用于下步反应。
步骤3:将步骤2所得中间体b的粗品溶于600毫升甲醇中并转移到加氢反应釜中,向反应体系中分别加入氢氧化钠1.1g,兰尼镍7.3克,封闭反应釜并先后用氮气、氢气各置换三次,最后保持釜内氢气压力3mpa到3.5mpa,反应温度在30分钟内由25℃升温至75℃,搅拌速度为500转/分钟,反应维持4小时直至反应完全。待反应体系将至室温,通过精馏的方式进行产品纯化,最终得到1,9-壬二胺68.5克,总收率为86.7%,其1h-nmr(300mhz,cdcl3)图谱与图1类似,确认为1,9-壬二胺。
- 还没有人留言评论。精彩留言会获得点赞!