具有1个烃基的异氰脲酸衍生物的制造方法与流程
本发明涉及具有1个烃基作为与氮原子结合的取代基的异氰脲酸衍生物的、新的制造方法。
背景技术:
一直以来已知异氰脲酸衍生物及其合成方法。例如,在非专利文献1的393页~396页中记载了与异氰脲酸单烷基酯有关的合成方法。进一步在非专利文献4中记载了与非专利文献1的394页的细节有关的内容,但在3618页中,关于具有ch3基和sec-c4h9基的异氰脲酸单烷基酯衍生物的合成在250℃的高温条件下进行反应。此外,在非专利文献2中介绍了2,4,6-三(苄氧基)1,3,5-三嗪、4,6-双(苄氧基)-1,3,5-三嗪-2,4(1h,3h)-二酮和6-(苄氧基)-1,3,5-三嗪-2,4(1h,3h)-二酮之中的6-(苄氧基)-1,3,5-三嗪-2,4(1h,3h)-二酮作为苄基化试剂显示最优异的反应性的研究结果。在非专利文献3中记载了使用了四丁基异氰脲酸铵的n-甲基化,异氰脲酸单甲酯、异氰脲酸二甲酯、和异氰脲酸三甲酯都生成了。即,在本非专利文献3所记载的方法中,不能避免这3种n-甲基化异氰脲酸酯的混合物非选择性地生成。
异氰脲酸衍生物用于各种用途。例如,专利文献1中记载了包含异氰脲酸衍生物的、光刻用防反射膜形成用组合物。专利文献2中记载了包含使异氰脲酸衍生物与其它单体聚合而获得的聚合物的、粘接剂组合物。
现有技术文献
非专利文献
非专利文献1:edwinm.smolin;lorencerapoport.“isocyanuricacidandderivatives”.thechemistryofheterocycliccompounds.s-triazinesandderivatives.,intersciencepublishers,inc.,pp.389-422(1959)
非专利文献2:journaloforganicchemistry,80,pp.11200-11205(2015)
非专利文献3:tetrahedronletters,44,pp.4399-4402(2003)
非专利文献4:journalofamericanchemicalsociety,75,pp.3617-3618(1953)
专利文献
专利文献1:国际公开wo02/086624号
专利文献2:国际公开wo2013/035787号
技术实现要素:
发明所要解决的课题
异氰脲酸衍生物之中,关于具有1个烷基的异氰脲酸二烷基酯,以往已知的制造方法也具有下述问题:需要在150℃以上的高温下进行长时间的加热,反应产物的选择性低等,工业上不可以说有用,此外从安全性的观点考虑具有令人担心的工序。
用于解决课题的方法
本发明的发明人发现,以下述式(0)所示的化合物,例如比较便宜且容易获得的氰尿酰氯(别名:2,4,6-三氯-1,3,5-三嗪)作为起始原料,经过获得第一中间体的工序、获得第二中间体的工序、和获得第三中间体的工序,最终获得具有1个烃基的异氰脲酸衍生物的制造方法。即,本发明是具有1个烃基的异氰脲酸衍生物的制造方法,其包含下述工序:由下述式(0)所示的化合物获得下述式(1)所示的化合物的第一工序;由上述式(1)所示的化合物获得下述式(2)所示的化合物的第二工序;由上述式(2)所示的化合物获得下述式(3)所示的化合物的第三工序;以及由上述式(3)所示的化合物获得下述式(4)所示的化合物的第四工序,
(式中,x1分别表示氯原子、氟原子或溴原子,bn表示苯环的至少1个氢原子可以被甲基取代的苄基,r表示碳原子数1~10的烃基。)
全部工序都在不超过100℃的温度下进行。
上述碳原子数1~10的烃基为例如烷基。
上述第一工序为通过使上述式(0)所示的化合物、与苯环的至少1个氢原子可以被甲基取代的苄醇在选自叔胺、碱金属碳酸盐和碱金属碳酸氢盐中的碱的存在下反应,将所获得的反应产物使用醇类进行洗涤,由此获得上述式(1)所示的化合物的工艺。相对于上述式(0)所示的化合物1.0摩尔当量,可以分别使用例如2.7摩尔当量~5.0摩尔当量的上述苯环的至少1个氢原子可以被甲基取代的苄醇和上述碱。
作为上述第一工序中使用的碱,可以选择叔胺,作为该叔胺,可举出例如二异丙基乙基胺。作为上述碱,代替叔胺,也可以使用碱金属碳酸盐或碱金属碳酸氢盐。作为该碱金属碳酸盐,可举出例如碳酸钠,作为该碱金属碳酸氢盐,可举出例如碳酸氢钠。作为上述苯环的至少1个氢原子可以被甲基取代的苄醇,可举出例如苄醇和4-甲基苯甲醇。
作为上述第一工序中用于对反应产物进行洗涤的醇类,可举出例如乙醇。
上述第二工序为通过使上述式(1)所示的化合物在包含选自n-甲基吗啉、n-乙基吗啉、n-甲基哌啶、n-乙基哌啶、n-甲基吡咯烷和n-乙基吡咯烷中的杂环式化合物、和乙酸或甲酸的溶液中反应,将所获得反应产物使用选自酯类、芳香族烃类、醇类和醚类中的至少1种溶剂进行洗涤,来获得上述式(2)所示的化合物的工艺。上述杂环式化合物具有至少1个氮原子作为杂原子,并且在n位具有取代基。
作为上述第二工序中用于对反应产物进行洗涤的至少1种溶剂,可以选择芳香族烃类,作为该芳香族烃类,可举出例如甲苯。作为该至少1种溶剂,也可以选择酯类、醇类或醚类,作为该酯类,可举出例如乙酸乙酯,作为该醇类,可举出例如乙醇,作为该醚类,可举出例如环戊基甲基醚。
上述第三工序为通过使上述式(2)所示的化合物、与具有碳原子数1~10的烃基的下述式(5)、式(6)、式(7)或式(8)所示的化合物在碱金属碳酸盐的存在下反应,将所获得的反应产物使用选自醇类和醚类中的至少1种溶剂进行洗涤,来获得上述式(3)所示的化合物的工艺。
(式中,r表示碳原子数1~10的烃基,x2表示卤原子。)
上述第三工序中使用的式(5)、式(6)、式(7)或式(8)所示的化合物为例如选自烷基卤化物、甲苯磺酸烷基酯、甲磺酸烷基酯和硫酸二烷基酯中的烷基化剂。
作为上述烷基化剂,可以选择烷基卤化物或硫酸二烷基酯。作为该烷基卤化物,可举出例如甲基碘、乙基溴、丙基溴、烯丙基溴、和炔丙基溴,作为该硫酸二烷基酯,可举出例如硫酸二甲酯。作为上述烷基化剂,代替烷基卤化物和硫酸二烷基酯,也可以使用甲苯磺酸烷基酯或甲磺酸烷基酯。作为该甲苯磺酸烷基酯,可举出例如对甲苯磺酸甲酯和对甲苯磺酸乙酯,作为该甲磺酸烷基酯,可举出例如甲磺酸乙酯。
此外,作为上述第三工序中使用的碱金属碳酸盐,可举出例如碳酸钾和碳酸铯。
进一步,作为上述第三工序中用于对反应产物进行洗涤的至少1种溶剂,可以选择醇类,作为该醇类,可举出例如乙醇。作为该至少1种溶剂,也可以选择醚类,作为该醚类,可举出例如环戊基甲基醚。
上述第四工序为通过使上述式(3)所示的化合物与醇化合物在三氟甲磺酸或三氟甲磺酸三甲基甲硅烷基酯的存在下反应,将所获得的反应产物使用选自酯类、烷基卤化物类和醇类中的至少1种溶剂进行洗涤,来获得上述式(4)所示的化合物的工艺。
作为上述第四工序中使用的醇化合物,可举出例如甲醇。
作为上述第四工序中用于对反应产物进行洗涤的至少1种溶剂,可以选择酯类,作为该酯类,可举出例如乙酸乙酯。作为该至少1种溶剂,也可以选择烷基卤化物类或醇类,作为该烷基卤化物类,可举出例如氯仿,作为该醇类,可举出例如乙醇。
上述第一工序~第四工序在不超过100℃的温度下进行。所谓该不超过100℃的温度,为0℃~100℃,为例如0℃~50℃。
发明效果
本发明涉及的具有1个烃基的异氰脲酸衍生物的制造方法,在全部工序中,不存在在超过100℃的温度下进行的工序,而且在对反应产物进行纯化时不需要采用柱色谱的纯化,因此工业上有用。进一步,通过本发明,可以与以往相比以高纯度(纯度98%以上)获得作为目标的具有1个烃基的异氰脲酸衍生物。
附图说明
图1是表示将第一工序中获得的实施例1的化合物通过高效液相色谱测定而得的结果的色谱图。
图2是表示将第二工序中获得的实施例4的化合物通过高效液相色谱测定而得的结果的色谱图。
图3是表示将第三工序中获得的实施例7的化合物通过高效液相色谱测定而得的结果的色谱图。
图4是表示将第四工序中获得的实施例9的化合物通过高效液相色谱测定而得的结果的色谱图。
图5是表示将比较例16中获得的化合物通过高效液相色谱测定而得的结果的色谱图。
具体实施方式
本发明涉及的具有1个烃基的异氰脲酸衍生物的制造方法,通过包含上述第一工序~第四工序,来获得导入了1个烃基作为与异氰脲酸的氮原子结合的取代基的上述式(4)所示的化合物。在该式(4)中,r表示被导入的碳原子数1~10的烃基。该烃基可以为直链状、支链状、环状中的任一种,可以具有至少1个双键或三键。在上述烃基为烷基的情况下,作为该烷基,可举出例如,甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、正己基、正庚基、正辛基、正戊基、正壬基、正癸基、环己基甲基、和环戊基甲基。作为除了烷基以外的上述烃基,可举出例如,苄基、烯丙基、和炔丙基。
在本发明的制造方法的第一工序中,使上述式(0)所示的化合物、与苯环的至少1个氢原子可以被甲基取代的苄醇在叔胺那样的碱的存在下反应。通过该反应,可以获得上述式(0)所示的化合物的3个取代基x1之中的2个取代基x1被取代成来源于上述苄醇的苄氧基的上述式(1)所示的化合物。例如,使用了氰尿酰氯作为上述式(0)所示的化合物时,通过上述反应,可以获得氰尿酰氯的3个氯原子之中的2个氯原子被取代成苄氧基的上述式(1)所示的化合物(式中,x1表示氯原子)。为了优先获得上述式(1)所示的化合物,相对于上述式(0)所示的化合物,例如氰尿酰氯1.0摩尔当量,优选分别使用2.7摩尔当量~5.0摩尔当量的上述苄醇、和叔胺等碱,更优选使用4.0摩尔当量~5.0摩尔当量。在相对于上述式(0)所示的化合物1.0摩尔当量,使上述苄醇、和叔胺等的碱分别以小于2.0摩尔当量使用而反应的情况下,与作为目标的上述式(1)所示的化合物相比,优先副生成上述式(0)所示的化合物的3个取代基x1之中的1个取代基x1被取代成来源于上述苄醇的苄氧基的化合物。因此,上述式(1)所示的化合物的收率降低。
作为上述反应所使用的叔胺,除了上述二异丙基乙基胺以外,还可举出三乙基胺、三丁基胺、和1,8-二氮杂二环[5.4.0]-7-十一碳烯作为优选例。此外,在溶液中进行上述反应的情况下,作为溶剂,优选为烷基卤化物类。作为上述烷基卤化物类,可举出例如,氯仿、二氯甲烷、四氯化碳、和二氯乙烷。
上述反应的细节从向将上述式(0)所示的化合物、例如氰尿酰氯和烷基卤化物类混合而得的溶液中滴加将苯环的至少1个氢原子可以被甲基取代的苄醇、叔胺等碱和烷基卤化物类混合而得的溶液开始。该滴加时的温度优选为0℃~5℃。原因是,在高于5℃的温度下滴加的情况下,由于与作为目标的上述式(1)所示的化合物一起,生成副产物,从而所得的上述式(1)所示的化合物的纯度和收率降低。滴加后的反应温度没有特别限定,通常为0℃~40℃,优选为20℃~30℃。反应时间通常为12小时~24小时,优选为15小时~18小时。
对通过上述反应而获得的反应产物进行分液操作,将该反应产物浓缩后,使用醇类进行洗涤来获得上述式(1)所示的化合物。作为上述洗涤所使用的醇类,除了上述乙醇以外,还可举出甲醇、异丙醇、正丙醇、仲丁醇、叔丁醇、正丁醇和环己醇作为优选例。上述洗涤所使用的溶剂的使用量相对于上述式(0)所示的化合物(例如,氰尿酰氯),优选为0.5质量倍~3.0质量倍,更优选为2.0质量倍。上述洗涤时的温度没有特别限定,通常为0℃~40℃,优选为0℃~5℃。洗涤时间通常为10分钟~1小时,优选为30分钟~1小时。
本发明的制造方法的第二工序中,使上述式(1)所示的化合物在选自n-甲基吗啉、n-乙基吗啉、n-甲基哌啶、n-乙基哌啶、n-甲基吡咯烷和n-乙基吡咯烷中的杂环式化合物和乙酸或甲酸的存在下反应。此外,在溶液中进行反应的情况下,作为溶剂,优选为醇类。作为上述醇类,除了上述甲醇以外,还可举出例如,乙醇、异丙醇、正丙醇、仲丁醇、叔丁醇、正丁醇和环己醇。
上述反应的细节从向将选自n-甲基吗啉、n-乙基吗啉、n-甲基哌啶、n-乙基哌啶、n-甲基吡咯烷和n-乙基吡咯烷中的杂环式化合物、乙酸或甲酸、以及醇类混合而得的溶液中加入上述式(1)所示的化合物开始。该加入时的温度优选为0℃~5℃。加入后的反应温度没有特别限定,通常为0℃~40℃,优选为20℃~30℃。反应时间通常为30分钟~3小时,优选为1小时~2小时。
对通过上述反应而获得的反应产物进行分液操作,将该反应产物浓缩后,使用选自酯类、芳香族烃类、醇类和醚类中的至少1种溶剂进行洗涤,来获得上述式(2)所示的化合物。作为上述洗涤所使用的溶剂,在选择酯类情况下,除了上述乙酸乙酯以外,还可举出乙酸甲酯、乙酸丁酯和丙酸甲酯作为优选例,在选择芳香族烃类的情况下,除了上述甲苯以外,还可举出苯、二甲苯、均三甲苯、氯苯、二氯苯、硝基苯、和四氢化萘作为优选例,在选择醇类的情况下,除了上述乙醇以外,还可举出甲醇、异丙醇、正丙醇、仲丁醇、叔丁醇、正丁醇和环己醇作为优选例,在选择醚类的情况下,除了上述环戊基甲基醚以外,还可举出乙醚、二异丙基醚(日文原文:ジイソプロピルピルエーテル)、甲基叔丁基醚、四氢呋喃、和二
本发明的制造方法的第三工序中,使上述式(2)所示的化合物、与上述式(5)、式(6)、式(7)或式(8)所示的化合物在碱金属碳酸盐的存在下反应。上述式(5)、式(6)、式(7)或式(8)所示的化合物的使用量相对于上述式(2)所示的化合物1.0摩尔当量,优选为1.0摩尔当量~1.5摩尔当量,更优选为1.25摩尔当量。上述碱金属碳酸盐的使用量相对于上述式(2)所示的化合物1.0摩尔当量,优选为1.0摩尔当量~1.5摩尔当量,更优选为1.25摩尔当量。上述反应时的温度没有特别限定,通常为0℃~40℃,优选为20℃~30℃。反应时间通常为30分钟~2小时,优选为30分钟~1小时。在溶液中进行上述反应的情况下,作为溶剂,优选使用选自非质子性极性溶剂和芳香族烃类中的至少1种溶剂。作为上述非质子性极性溶剂,可举出例如,二甲亚砜、n-甲基吡咯烷酮、二甲基乙酰胺、和二甲基甲酰胺,在选择芳香族烃类的情况下,可举出甲苯、苯、二甲苯、均三甲苯、氯苯、二氯苯、硝基苯、和四氢化萘作为优选例。
通过选择本发明的制造方法的第三工序中使用的上述式(5)、式(6)、式(7)或式(8)所示的化合物,来确定作为与异氰脲酸的氮原子结合的取代基而被导入的烃基的种类。例如,作为上述式(5)、式(6)、式(7)或式(8)所示的化合物,通过使用具有甲基的烷基化剂来导入甲基,通过使用具有乙基的烷基化剂来导入乙基。
对通过上述反应而获得的反应产物进行分液操作,将该反应产物浓缩后,通过使用选自醇类和醚类中的至少1种溶剂进行洗涤,来获得上述式(3)所示的化合物。作为上述洗涤所使用的溶剂,在选择醇类的情况下,除了上述乙醇以外,还可举出甲醇、异丙醇、正丙醇、-丁基醇、叔丁醇、正丁醇和环己醇作为优选例,在选择醚类的情况下,除了上述环戊基甲基醚以外,还可举出乙醚、二异丙基醚(日文原文:ジイソプロピルピルエーテル)、甲基叔丁基醚、四氢呋喃、和二
本发明的制造方法的第四工序中,使上述式(3)所示的化合物与醇化合物在三氟甲磺酸或三氟甲磺酸三甲基甲硅烷基酯的存在下反应。作为上述醇化合物,除了上述甲醇以外,还可举出例如乙醇、异丙醇、正丙醇、仲丁醇、叔丁醇、正丁醇、环己醇、和苯酚。上述醇化合物的使用量相对于上述式(3)所示的化合物1.0摩尔当量,优选为2摩尔当量~3摩尔当量,更优选为2.4摩尔当量。上述三氟甲磺酸或三氟甲磺酸三甲基甲硅烷基酯的使用量相对于上述式(3)所示的化合物1.0摩尔当量,优选为0.7摩尔当量~1.0摩尔当量,更优选为1.0摩尔当量。在以小于0.7摩尔当量使用而进行反应的情况下,副生出与作为目标的上述式(4)所示的化合物不同的杂质。因此,上述式(4)所示的化合物的纯度和收率降低。上述反应时的温度没有特别限定,通常为0℃~40℃,优选为20℃~30℃。反应时间通常为1小时~5小时,优选为1小时~2小时。在溶液中进行上述反应的情况下,作为溶剂,优选为醚类和芳香族烃类。作为上述醚类,可举出例如,乙醚、二异丙基醚(日文原文:ジイソプロピルピルエーテル)、甲基叔丁基醚、环戊基甲基醚、四氢呋喃、和二
在通过上述反应而获得的反应产物中加入有机碱进行浓缩后,使用选自酯类、烷基卤化物类和醇类中的至少1种溶剂进行洗涤,来获得上述式(4)所示的化合物。作为上述有机碱,可举出吡啶、4-二甲基氨基吡啶、三乙基胺、三丁基胺、n,n-二甲基苯胺、和1,8-二氮杂二环[5.4.0]-7-十一碳烯作为优选例。上述有机碱的使用量相对于上述式(3)所示的化合物1.0摩尔当量,优选为1.0摩尔当量~2.0摩尔当量,更优选为1.0摩尔当量。作为上述洗涤所使用的溶剂,在选择酯类的情况下,除了上述乙酸乙酯以外,还可举出乙酸甲酯、乙酸丁酯和丙酸甲酯作为优选例,在选择烷基卤化物类的情况下,除了上述氯仿以外,还可举出二氯甲烷、四氯化碳和二氯乙烷作为优选例,在选择醇类的情况下,除了上述乙醇以外,还可举出甲醇、异丙醇、正丙醇、仲丁醇、叔丁醇、正丁醇和环己醇作为优选例。上述洗涤所使用的溶剂的使用量相对于上述式(3)所示的化合物,优选为2.0质量倍~5.0质量倍,更优选为3.0质量倍。上述洗涤时的温度没有特别限定,通常为0℃~40℃,优选为20℃~30℃。洗涤时间通常为10分钟~1小时,优选为10分钟~30分钟。
实施例
以下,举出具体例说明本发明涉及的具有1个烃基的异氰脲酸衍生物的制造方法。然而,本发明不限定于以下举出的具体例。
[hplc分析条件-1]
后述的例子所示的纯度为由高效液相色谱(以下,简称为hplc。)得到的测定结果,测定条件等如下所述。该纯度是将各成分的峰如图1所示那样分割,将各个峰的面积值以百分率算出的值。
装置:(株)日立ハイテクノロジーズ制,l2000系列
柱:xbridge〔注册商标〕behc18column,
洗脱液:乙腈/0.2%乙酸铵水溶液=8/2(v/v)
流量:1.0ml/分钟
检测器:uv(254nm)
柱温度:40℃
分析时间:15分钟
进样量:2.0μl
稀释溶剂:乙腈
[hplc分析条件-2]
后述的例子所示的纯度为由hplc得到的测定结果,测定条件等如下所述。该纯度是将各成分的峰如图2、图3和图4所示那样分割,将各个面积值以百分率算出的值。
装置:(株)岛津制作所制,lc-2010a
柱:xbridge〔注册商标〕behc18column,
洗脱液:乙腈/0.2%乙酸铵水溶液=3/7(v/v)(0分钟~5分钟),从3/7(v/v)向8/2(v/v)变更组成比(5分钟~10分钟),8/2(v/v)(10分钟~15分钟)
流量:1.0ml/分钟
检测器:uv(210nm)
柱温度:40℃
分析时间:25分钟
进样量:1.0μl
稀释溶剂:乙腈/水=1/1(w/w)
[收率算出方法]
后述的例子所示的收率为使用所得的化合物的重量与理论收量以百分率算出的值。另外,上述理论收量是通过将合成所使用的原料化合物的摩尔数与所得的化合物的分子量相乘而算出的值。
[第一工序]
<合成例1>
将氰尿酰氯(东京化成工业(株)制)20.00g和氯仿120.00g混合,一边搅拌一边冷却直到0℃。向其中滴加将苄醇(关东化学(株)制)46.91g、二异丙基乙基胺56.07g和氯仿60.00g混合而得的溶液。在滴加结束后,升温直到25℃并进行17小时搅拌,向反应溶液加入饱和nh4cl水溶液200.00g,进行了分液。接着,在有机层中加入饱和食盐水200.00g,将该分液操作重复2次。将有机层在减压下蒸馏除去溶剂,然后将残渣在40℃下减压干燥,从而作为粗品(粗生成物)而获得了上述式(1a)所示的三嗪化合物57.56g(粗品收率>99.9%)。另外,在上述hplc分析条件-1下对所得的化合物进行测定,结果纯度为61.4%。
<实施例1>
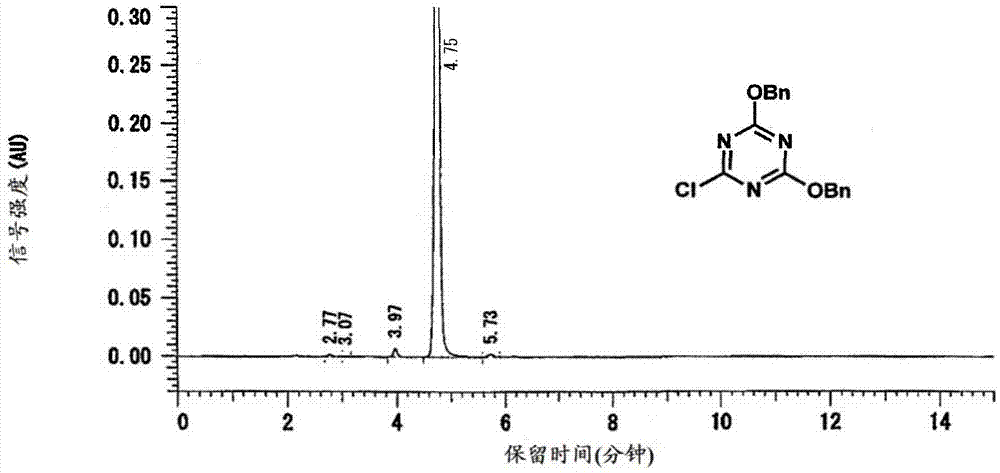
在合成例1中获得的三嗪化合物的粗品3.00g中加入乙醇2.08g,在25℃下进行10分钟搅拌。搅拌后,进行过滤,进一步用乙醇1.04g进行了2次滤饼洗涤。这里,所谓滤饼,表示通过将浆料等固液混合物过滤来分离液体而残留的固体物质。将所得的结晶在40℃下减压干燥,从而作为淡黄色固体而获得了上述式(1a)所示的三嗪化合物1.30g(收率70.4%)。另外,在上述hplc分析条件-1下对所得的化合物进行测定,结果纯度为98.4%。将通过该测定而获得的色谱图示于图1中。
<实施例2>
在合成例1中获得的三嗪化合物的粗品3.00g中加入乙醇2.08g,在0℃下进行10分钟搅拌,除此以外,与实施例1同样地实施,结果作为淡黄色固体而获得了上述式(1a)所示的三嗪化合物1.49g(收率80.4%)。另外,在上述hplc分析条件-1下对所得的化合物进行测定,结果纯度为98.2%。
<比较例1>
在合成例1中获得的三嗪化合物的粗品3.00g中加入甲苯2.08g,除此以外,与实施例1同样地进行。搅拌后,完全溶解,因此得不到结晶。
<比较例2>
在合成例1中获得的三嗪化合物的粗品3.00g中加入庚烷2.08g,滤饼洗涤溶剂使用了庚烷,除此以外,与实施例1同样地进行。作为淡黄色固体而获得了上述式(1a)所示的三嗪化合物1.63g(收率87.9%)。另外,在上述hplc分析条件-1下对所得的化合物进行测定,结果纯度为76.8%。
<比较例3>
在合成例1中获得的三嗪化合物的粗品3.00g中加入乙酸乙酯/庚烷=1/9(w/w)2.08g,滤饼洗涤溶剂使用了乙酸乙酯/庚烷=1/9(w/w),除此以外,与实施例1同样地进行。作为淡黄色固体而获得了上述式(1a)所示的三嗪化合物1.10g(收率59.6%)。另外,在上述hplc分析条件-1下对所得的化合物进行测定,结果纯度为86.1%。
<比较例4>
使用合成例1中获得的三嗪化合物的粗品3.00g,以乙酸乙酯/己烷=1/10(w/w)作为展开溶剂进行了硅胶柱色谱。将所得的溶液在40℃下浓缩和减压干燥,从而作为白色固体而获得了上述式(1a)所示的三嗪化合物1.58g(收率85.3%)。另外,在上述hplc分析条件-1下对所得的化合物进行测定,结果纯度为92.5%。
表1
[第二工序]
<合成例2>
将n-甲基吗啉(东京化成工业(株)制)30.86g、乙酸(关东化学(株)制)9.16g和甲醇250.00g混合,冷却直到0℃。向其中一边搅拌一边加入实施例1中获得的三嗪化合物25.00g。接着,升温直到25℃并进行1小时搅拌,向反应溶液加入乙酸9.16g,在减压下蒸馏除去溶剂。向其中加入氯仿250.00g与1mhcl250.00g进行了分液。进一步向有机层加入饱和食盐水250.00g进行了分液。将有机层在40℃下减压干燥,作为粗品(粗成生物)而获得了上述式(2)所示的三嗪-酮化合物22.98g(粗品收率97.4%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为88.7%。
<实施例3>
在合成例2中获得的三嗪-酮化合物的粗品3.00g中加入乙酸乙酯6.53g,在25℃下进行10分钟搅拌。搅拌后,进行过滤,进一步用乙酸乙酯3.26g进行了2次滤饼洗涤。将所得的结晶在40℃下减压干燥,从而作为白色固体而获得了上述式(2)所示的三嗪-酮化合物2.56g(收率83.3%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为95.8%。
<实施例4>
在合成例2中获得的三嗪-酮化合物的粗品3.00g中加入甲苯6.53g,滤饼洗涤溶剂使用了甲苯,除此以外,与实施例3同样地进行。作为白色固体而获得了上述式(2)所示的三嗪-酮化合物2.66g(收率86.4%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为96.0%。将通过该测定而获得的色谱图示于图2中。
<实施例5>
在合成例2中获得的三嗪-酮化合物的粗品3.00g中加入乙醇6.53g,滤饼洗涤溶剂使用了乙醇,除此以外,与实施例3同样地进行。作为白色固体而获得了上述式(2)所示的三嗪-酮化合物2.54g(收率82.4%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为96.1%。
<实施例6>
在合成例2中获得的三嗪-酮化合物的粗品3.00g中加入环戊基甲基醚6.53g,滤饼洗涤溶剂使用了环戊基甲基醚,除此以外,与实施例3同样地进行。作为白色固体而获得了上述式(2)所示的三嗪-酮化合物2.64g(收率85.8%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为95.9%。
<比较例5>
在合成例2中获得的三嗪-酮化合物的粗品3.00g中加入庚烷6.53g,滤饼洗涤溶剂使用了庚烷,除此以外,与实施例3同样地进行。作为白色固体而获得了上述式(2)所示的三嗪-酮化合物2.73g(收率88.7%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为92.8%。
<比较例6>
在合成例2中获得的三嗪-酮化合物的粗品3.00g中加入氯仿6.53g,滤饼洗涤溶剂使用了氯仿,除此以外,与实施例3同样地进行。作为白色固体而获得了上述式(2)所示的三嗪-酮化合物1.88g(收率61.0%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为96.6%。
<比较例7>
使用合成例2中获得的三嗪-酮化合物的粗品3.00g,以氯仿/乙酸乙酯=4/1(w/w)作为展开溶剂进行了硅胶柱色谱。将所得的溶液在40℃下浓缩和减压干燥,从而作为白色固体而获得了上述式(2)所示的三嗪-酮化合物2.42g(收率78.6%)。另外,在上述hplc分析条件-1下对所得的化合物进行测定,结果纯度为99.7%。
表2
[第三工序]
<合成例3>
将实施例4中获得的三嗪-酮化合物21.00g、碳酸铯(东京化成工业(株)制)27.65g和二甲亚砜210.00g混合,在25℃下搅拌。向其中滴加甲基碘(东京化成工业(株)制)12.05g。滴加结束后,在25℃下进行1小时搅拌,向反应溶液加入甲苯210.00g,进行了过滤。接着,在滤液中加入水210.00g,进行了分液。将该分液操作重复2次。将有机层在减压下蒸馏除去溶剂后,将残渣在40℃下减压干燥,从而作为粗品(粗成生物)而获得了上述式(3a)所示的单甲基三嗪-酮化合物20.89g(粗品收率95.2%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为85.6%。
<实施例7>
在合成例3中获得的单甲基三嗪-酮化合物的粗品3.00g中加入乙醇6.03g,在25℃下进行10分钟搅拌。搅拌后,进行过滤,进一步用乙醇3.02g进行了2次滤饼洗涤。将所得的结晶在40℃下减压干燥,从而作为白色固体而获得了上述式(3a)所示的单甲基三嗪-酮化合物2.30g(收率73.0%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为98.0%。此外,测定了该化合物的1hnmr(500mhz,cdcl3),结果为δ7.48-7.34(m,10h),5.48(s,2h),5.44(s,2h),3.41(s,3h)。将通过该测定而获得的色谱图示于图3中。
<实施例8>
在合成例3中获得的单甲基三嗪-酮化合物的粗品3.00g中加入环戊基甲基醚6.03g,滤饼洗涤溶剂使用了环戊基甲基醚,除此以外,与实施例7同样地进行。作为白色固体而获得了上述式(3a)所示的单甲基三嗪-酮化合物2.30g(收率72.9%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为98.5%。
<比较例8>
在合成例3中获得的单甲基三嗪-酮化合物的粗品3.00g中加入庚烷6.03g,滤饼洗涤溶剂使用了庚烷,除此以外,与实施例7同样地进行。作为白色固体而获得了上述式(3a)所示的单甲基三嗪-酮化合物2.77g(收率87.8%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为89.8%。
<比较例9>
在合成例3中获得的单甲基三嗪-酮化合物的粗品3.00g中加入乙酸乙酯6.03g,滤饼洗涤溶剂使用了乙酸乙酯,除此以外,与实施例7同样地进行。作为白色固体而获得了上述式(3a)所示的单甲基三嗪-酮化合物1.69g(收率53.6%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为98.7%。
<比较例10>
在合成例3中获得的单甲基三嗪-酮化合物的粗品3.00g中加入甲苯6.03g,滤饼洗涤溶剂使用了甲苯,除此以外,与实施例7同样地进行。作为白色固体而获得了上述式(3a)所示的单甲基三嗪-酮化合物1.57g(收率49.8%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为98.4%。
<比较例11>
在合成例3中获得的单甲基三嗪-酮化合物的粗品3.00g中加入了氯仿6.03g,除此以外,与实施例7同样地进行。搅拌后,完全溶解,因此得不到结晶。
<比较例12>
使用合成例3中获得的单甲基三嗪-酮化合物的粗品3.00g,作为展开溶剂从氯仿/庚烷=7/3(w/w)向氯仿/庚烷=9/1(w/w)变更组成,进一步向氯仿/乙酸乙酯=9/1(w/w)变更组成而进行了硅胶柱色谱。将所得的溶液在40℃下浓缩和减压干燥,从而作为白色固体而获得了上述式(3a)所示的三嗪-酮化合物2.82g(收率89.5%)。另外,在上述hplc分析条件-2下对所得的化合物测定,结果纯度为91.4%。
表3
[第四工序]
<实施例9>
将实施例7中获得的单甲基三嗪-酮化合物2.00g、1,4-二
<实施例10>
作为洗涤溶剂使用了氯仿,除此以外,与实施例9同样地进行,结果作为白色固体而获得了上述式(4a)所示的异氰脲酸单甲酯0.81g(收率91.4%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为98.2%。
<实施例11>
使用了乙醇作为洗涤溶剂,除此以外,与实施例9同样地进行,结果作为白色固体而获得了上述式(4a)所示的异氰脲酸单甲酯0.67g(收率75.6%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为98.2%。
<比较例13>
使用了甲苯作为洗涤溶剂,除此以外,与实施例9同样地进行,结果作为白色固体而获得了上述式(4a)所示的异氰脲酸单甲酯0.91g(收率102.1%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为97.2%。
<比较例14>
使用了庚烷作为洗涤溶剂,除此以外,与实施例9同样地进行,结果作为白色固体而获得了上述式(4a)所示的异氰脲酸单甲酯1.68g(收率189.1%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为95.6%。
<比较例15>
使用了环戊基甲基醚作为洗涤溶剂,除此以外,与实施例9同样地进行,结果作为白色固体而获得了上述式(4a)所示的异氰脲酸单甲酯0.92g(收率103.2%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为96.3%。
表4
实施例9~实施例11的结果显示出能够以高纯度(98%以上)获得具有1个烃基的异氰脲酸衍生物。这样的异氰脲酸衍生物的制造方法不含在超过100℃的温度下进行的工序、和采用柱色谱的纯化工序,因此工业上是有用的。
<比较例16>
将实施例4中获得的三嗪-酮化合物100.00g、碳酸铯(东京化成工业(株)制)131.67g和n-甲基-2-吡咯烷酮1000.0g混合,在40℃下搅拌30分钟。如果,冷却直到小于5℃,滴加炔丙基溴(东京化成工业(株)制)48.07g。滴加结束后,在小于5℃下进行6小时搅拌,向反应溶液加入甲苯1000.0g,进行了过滤。接着,在滤液中加入水1000.0g,进行了分液。将该分液操作重复3次。向该溶液滴加三氟甲磺酸(东京化成工业(株)制)14.55g和甲醇24.86g。滴加结束后,在25℃下进行2小时搅拌,向反应溶液加入三乙基胺39.26g。在减压下蒸馏除去溶剂后,将残渣在40℃下减压干燥。接着,加入乙酸乙酯300.00g,在25℃下进行10分钟搅拌。搅拌后,进行过滤,进一步用乙酸乙酯100.00g进行了2次滤饼洗涤。将所得的结晶在40℃下减压干燥,从而作为白色固体而获得了上述式(4b)所示的异氰脲酸单炔丙酯14.26g(收率26.4%)。另外,在上述hplc分析条件-2下对所得的化合物进行测定,结果纯度为94.0%。此外,测定了该化合物的1hnmr(500mhz,dmso-d6),结果为δ11.56(s,2h),4.39(d,2h),3.20(t,1h)。将通过该测定而获得的色谱图示于图5中。本比较例是将没有进行使用了选自醇类和醚类中的至少1种溶剂的洗涤而获得的、包含上述式(3)所示的化合物的溶液适用于第四工序的例子。
产业可利用性
通过本发明制造的具有1个烃基的异氰脲酸衍生物例如可以适用于光刻用防反射膜形成用组合物、抗蚀剂下层膜形成用组合物、抗蚀剂上层膜形成用组合物、光固化性树脂组合物、热固性树脂组合物、平坦化膜形成用组合物、粘接剂组合物、其它组合物。
- 还没有人留言评论。精彩留言会获得点赞!