从100pg以下的人类基因组DNA判别其来源的方法、识别个人的方法及分析造血干细胞的植活程度的方法与流程
本发明涉及一种从100pg以下的人类基因组dna(deoxyribonucleicacid;脱氧核糖核酸)判别其来源的方法、识别个人的方法及分析造血干细胞的植活(engraftmen)程度的方法。
背景技术:
为了从微量的基因组dna获取碱基序列信息,需要通过全基因组扩增对成为分析对象的dna进行扩增之后,通过pcr(polymerasechainreaction;聚合酶链反应)对所希望的目标区域进行扩增。但是,该方法中,由于扩增区域的不均匀化或扩增错误等,有时无法获取高精度的碱基序列信息。
若不进行全基因组扩增而通过pcr直接对目标区域进行扩增,则能够排除上述的全基因组扩增的影响。但是,由于模板dna量极少,会产生过度的扩增反应引起的扩增错误或引物彼此的非特异性扩增(引物二聚体)等,有时无法获取准确的碱基序列信息。
作为抑制形成引物二聚体的方法,例如,专利文献1中记载有如下内容,即,针对引物的所有组合,计算表示引物之间的3’末端中的互补性的评分(3’末端的局部比对评分),选出引物彼此的互补性低的引物的组合,由此降低多重pcr中不同目标的引物彼此形成引物二聚体的可能性。
以往技术文献
专利文献
专利文献1:国际公开第2008/004691号
技术实现要素:
发明要解决的技术课题
但是,对极微量的模板dna的多重pcr中,考虑仅在引物端部进行退火而形成的引物二聚体也很重要,但专利文献1中记载的方法并未考虑至此。
因此,根据专利文献1中记载的引物的设计方法设计的引物中,对100pg以下的极微量的模板dna进行多重pcr时,未能对多个目标区域均匀地进行扩增。
因此,一直以来,很难从如从遗留dna或单细胞提取的dna的100pg以下的微量dna判别其来源、识别个人及分析造血干细胞的植活程度。
因此,本发明的课题在于,提供一种通过对100pg以下的人类基因组dna均匀地进行扩增,判别该100pg以下的人类基因组dna的来源的方法、识别个人的方法及分析造血干细胞的植活程度的方法。
用于解决技术课题的手段
本发明人等为了解决上述课题而反复进行深入研究,其结果,发现如下内容并完成了本发明:在将100pg以下的人类基因组dna作为模板进行多重pcr时使用的引物的设计方法中,对引物二聚体形成性进行评价,针对引物候选的碱基序列,在所比较的一部分序列包含引物的碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分,并根据所求出的局部比对评分进行第1阶段的选拔,针对包含引物候选的碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分,并根据所求出的总体比对评分进行第2阶段的选拔,若采用在第1阶段及第2阶段的任一阶段中均被选拔的引物,则抑制引物二聚体的形成,能够对多个目标区域均匀地进行扩增。
即,本发明提供以下的[1]至[6]。
[1]一种从100pg以下的人类基因组dna判别其来源的方法,其具备:
目标区域选择工序,从人类基因组dna上的区域选择用于获取碱基序列信息的至少1个目标区域;
dna提取工序,从人源试样提取人类基因组dna;
pcr扩增工序,从在上述dna提取工序中获得的人类基因组dna,将100pg以下的人类基因组dna作为模板,利用以对上述至少1个目标区域进行pcr扩增的方式设计的引物组合,对上述至少1个目标区域进行pcr扩增;
dna测序工序,解读在上述pcr扩增工序中获得的pcr扩增产物的dna碱基序列,获取上述至少1个目标区域的碱基序列信息;及
来源判别工序,根据上述碱基序列信息判别上述人类基因组dna的来源,
上述目标区域选择工序与上述dna提取工序为任意顺序,
上述从100pg以下的人类基因组dna判别其来源的方法中,
以对上述至少1个目标区域进行pcr扩增的方式设计的引物组合通过用于聚合酶链反应的引物组合的设计方法设计,该设计方法具备:
a)对象区域选择工序,从上述至少1个目标区域选择对象区域;
b)引物候选碱基序列制作工序,根据上述人类基因组dna上的上述对象区域两端的各个附近区域的碱基序列,分别各制作至少1个用于对上述对象区域进行pcr扩增的引物候选的碱基序列;
c)局部比对工序,针对从在上述引物候选碱基序列制作工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端这样的条件下,进行成对局部比对,从而求出局部比对评分;
d)第1阶段选拔工序,根据上述局部比对评分,进行用于对上述对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔;
e)总体比对工序,针对从在上述第1阶段选拔工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分;
f)第2阶段选拔工序,根据上述总体比对评分,进行用于对上述对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔;及
g)引物采用工序,采用在上述第1阶段选拔工序及上述第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述对象区域进行pcr扩增的引物的碱基序列,
上述局部比对工序及上述第1阶段选拔工序这两个工序和上述总体比对工序及上述第2阶段选拔工序这两个工序为任意顺序或同时进行。
[2]一种从100pg以下的人类基因组dna判别其来源的方法,其具备:
目标区域选择工序,从人类基因组dna上的区域选择用于获取碱基序列信息的至少1个目标区域;
dna提取工序,从人源试样提取人类基因组dna;
pcr扩增工序,从在上述dna提取工序中获得的人类基因组dna,将100pg以下的人类基因组dna作为模板,利用以对上述至少1个目标区域进行pcr扩增的方式设计的引物组合,对上述至少1个目标区域进行pcr扩增;
dna测序工序,解读在上述pcr扩增工序中获得的pcr扩增产物的dna碱基序列,获取上述至少1个目标区域的碱基序列信息;及
来源判别工序,根据上述碱基序列信息判别上述人类基因组dna的来源,
上述目标区域选择工序和上述dna提取工序为任意顺序,
上述从100pg以下的人类基因组dna判别其来源的方法中,
以对上述至少1个目标区域进行pcr扩增的方式设计的引物组合通过用于聚合酶链反应的引物组合的设计方法设计,该设计方法具备:
a1)对象区域选择第1工序,从上述至少1个目标区域选择第1对象区域;
b1)引物候选碱基序列制作第1工序,根据上述人类基因组dna上的上述第1对象区域的两端的各个附近区域的碱基序列,分别各制作至少1个用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列;
c1)局部比对第1工序,针对从在上述引物候选碱基序列制作第1工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分;
d1)第1阶段选拔第1工序,根据上述局部比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔;
e1)总体比对第1工序,针对从在上述第1阶段选拔第1工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分;
f1)第2阶段选拔第1工序,根据上述总体比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔;
g1)引物采用第1工序,采用在上述第1阶段选拔第1工序及上述第2阶段选拔第1工序二者中均被选拔的引物候选的碱基序列作为用于对上述第1对象区域进行pcr扩增的引物的碱基序列;
a2)对象区域选择第2工序,从上述至少1个目标区域中还未被选择的目标区域选择第2对象区域;
b2)引物候选碱基序列制作第2工序,根据上述人类基因组dna上的上述第2对象区域的两端的各个附近区域的碱基序列,分别各制作至少1个用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列;
c2)局部比对第2工序,针对从在上述引物候选碱基序列制作第2工序中制作的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分;
d2)第1阶段选拔第2工序,根据上述局部比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔;
e2)总体比对第2工序,针对从在上述第1阶段选拔第2工序中选拔的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分;
f2)第2阶段选拔第2工序,根据上述总体比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔;及
g2)引物采用第2工序,采用在上述第1阶段选拔第2工序及上述第2阶段选拔第2工序二者中均被选拔的引物候选的碱基序列作为用于对上述第2对象区域进行pcr扩增的引物的碱基序列,
上述局部比对第1工序及上述第1阶段选拔第1工序这两个工序和上述总体比对第1工序及上述第2阶段选拔第1工序这两个工序为任意顺序或同时进行,
上述局部比对第2工序及上述第1阶段选拔第2工序这两个工序和上述总体比对第2工序及上述第2阶段选拔第2工序这两个工序为任意顺序或同时进行,
在上述至少1个目标区域包含3个以上的目标区域的情况下,且采用用于对还未从上述3个以上的目标区域选择的第3及其后的对象区域进行pcr扩增的引物的碱基序列时,针对上述第3及其后的对象区域,反复进行上述对象区域选择第2工序至上述引物采用第2工序的各工序。
[3]一种从100pg以下的人类基因组dna判别其来源的方法,其具备:
目标区域选择工序,从人类基因组dna上的区域选择用于获取碱基序列信息的至少1个目标区域;
dna提取工序,从人源试样提取人类基因组dna;
pcr扩增工序,从在上述dna提取工序中获得的人类基因组dna,将100pg以下的人类基因组dna作为模板,利用以对上述至少1个目标区域进行pcr扩增的方式设计的引物组合,对上述至少1个目标区域进行pcr扩增;
dna测序工序,解读在上述pcr扩增工序中获得的pcr扩增产物的dna碱基序列,获取上述至少1个目标区域的碱基序列信息;及
来源判别工序,根据上述碱基序列信息判别上述人类基因组dna的来源,
上述目标区域选择工序和上述dna提取工序为任意顺序,
上述从100pg以下的人类基因组dna判别其来源的方法中,
以对上述至少1个目标区域进行pcr扩增的方式设计的引物组合通过用于聚合酶链反应的引物组合的设计方法设计,该设计方法具备:
a-0)对象区域多个选择工序,从上述至少1个目标区域选择多个对象区域;
b-0)引物候选碱基序列多个制作工序,根据上述人类基因组dna上的上述多个对象区域的各两端的各个附近区域的碱基序列,分别各制作至少1个用于分别对上述多个对象区域进行pcr扩增的引物候选的碱基序列;
c-1)第1局部比对工序,针对从在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列中用于对第1对象区域进行pcr扩增的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分;
d-1)第一第1阶段选拔工序,根据上述局部比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔;
e-1)第1总体比对工序,针对从在上述第一第1阶段选拔工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分;
f-1)第一第2阶段选拔工序,根据上述总体比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔;
g-1)第1引物采用工序,采用在上述第一第1阶段选拔工序及上述第一第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述第1对象区域进行pcr扩增的引物的碱基序列;
c-2)第2局部比对工序,针对从在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列中用于对第2对象区域进行pcr扩增的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分;
d-2)第二第1阶段选拔工序,根据上述局部比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔;
e-2)第2总体比对工序,针对从在上述第二第1阶段选拔工序中选拔的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分;
f-2)第二第2阶段选拔工序,根据上述总体比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔;及
g-2)第2引物采用工序,采用在上述第二第1阶段选拔工序及上述第二第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述第2对象区域进行pcr扩增的引物的碱基序列,
其中,上述第1局部比对工序及上述第一第1阶段选拔工序这两个工序和上述第1总体比对工序及上述第一第2阶段选拔工序这两个工序为任意顺序或同时进行,
上述第2局部比对工序及上述第二第1阶段选拔工序这两个工序和上述第2总体比对工序及上述第二第2阶段选拔工序这两个工序为任意顺序或同时进行,
在上述至少1个目标区域包含3个以上的目标区域,在上述对象区域多个选择工序中选择3个以上的对象区域,且在上述引物候选碱基序列多个制作工序中制作用于分别对上述3个以上的对象区域进行pcr扩增的引物候选的碱基序列的情况下,采用用于对第3及其后的对象区域进行pcr扩增的引物的碱基序列时,针对上述第3及其后的对象区域,反复进行上述第2局部比对工序至上述第2引物采用工序的各工序。
[4]根据上述[1]至[3]中任一项所述的方法,上述目标区域包含单核苷酸多态性和/或串联重复序列。
[5]一种识别个人的方法,其利用上述[1]至[4]中任一项所述的从100pg以下的人类基因组dna判别其来源的方法。
[6]一种分析造血干细胞的植活程度的方法,其利用上述[1]至[4]中任一项所述的从100pg以下的人类基因组dna判别其来源的方法。
发明效果
根据本发明,能够提供一种通过对100pg以下的人类基因组dna均匀地进行扩增,判别该100pg以下的人类基因组dna的来源的方法、识别个人的方法及分析造血干细胞的植活程度的方法。
根据本发明,即使是100pg以下的微量的dna量,也能够精度良好地进行dna分析。而且,即使是50pg以下的dna量,也能够实现高精度的dna分析,在人类基因组dna的来源的判别、识别个人的方法及造血干细胞的植活程度的分析方面有用。
附图说明
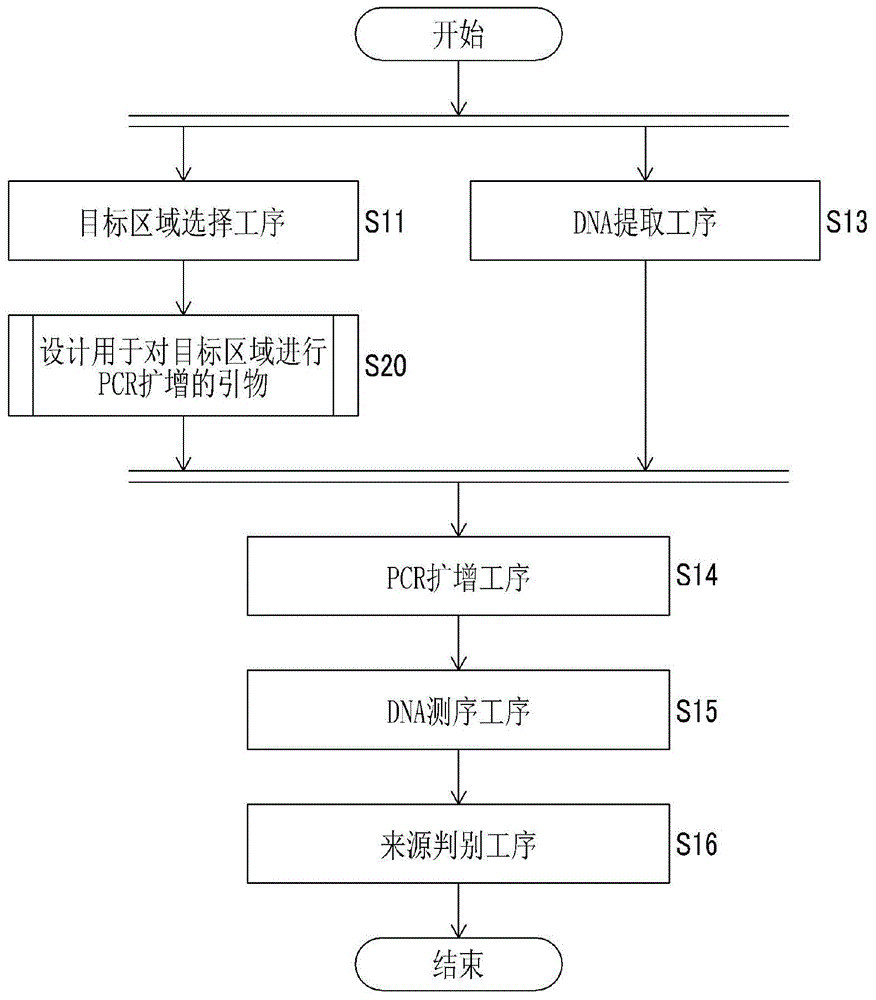
图1是表示本发明的从100pg以下的人类基因组dna判别其来源的方法的概略的流程图。
图2是说明用于聚合酶链反应的引物组合的设计方法的第1方式的流程图,该设计方法用于设计在本发明的从100pg以下的人类基因组dna判别其来源的方法中使用的以对目标区域进行pcr扩增的方式设计的引物组合。
图3是说明用于聚合酶链反应的引物组合的设计方法的第2方式的流程图,该设计方法用于设计在本发明的从100pg以下的人类基因组dna判别其来源的方法中使用的以对目标区域进行pcr扩增的方式设计的引物组合。
图4是说明用于聚合酶链反应的引物组合的设计方法的第2方式的流程图,该设计方法用于设计在本发明的从100pg以下的人类基因组dna判别其来源的方法中使用的以对目标区域进行pcr扩增的方式设计的引物组合。
图5是表示模板dna量(横轴)与序列读段的覆盖率成为19以上的区域的比例(纵轴)之间的关系的纵条形图。
图6是表示工作人员a及1~11(横轴)与snps信息[均质一致、异质不一致、ado(alleledropout;等位基因脱扣)、adi(alleledropin;等位基因插入)及均质不一致的个数]之间的关系的累积纵条形图。
图7是基于工作人员a及1~11的彼此之间的距离的系统树图。
具体实施方式
[从100pg以下的人类基因组dna判别其来源的方法]
本发明的从100pg以下的人类基因组dna判别其来源的方法(以下,有时简称为“本发明的判别来源的方法”。)具备:目标区域选择工序,从人类基因组dna上的区域选择用于获取碱基序列信息的至少1个目标区域;dna提取工序,从人源试样提取人类基因组dna;pcr扩增工序,从在上述dna提取工序中获得的人类基因组dna,将100pg以下的人类基因组dna作为模板,利用以对上述至少1个目标区域进行pcr扩增的方式设计的引物组合,对上述至少1个目标区域进行pcr扩增;dna测序工序,分析在上述pcr扩增工序中获得的pcr扩增产物的dna碱基序列,获取上述至少1个目标区域的碱基序列信息;及来源判别工序,根据上述碱基序列信息判别上述人类基因组dna的来源。
上述目标区域选择工序和上述dna提取工序为任意顺序。
在上述pcr扩增工序中为了通过聚合酶链反应对上述目标区域进行扩增而使用的引物组合通过后述的在“[用于聚合酶链反应的引物组合的设计方法]”中说明的引物组合的设计方法设计。
以下,对各工序进行详细说明。
<目标区域选择工序>
目标区域选择工序中,从人类基因组dna上的区域,选择获取用于判别作为多重pcr的模板dna的、100pg以下的人类基因组dna的来源的碱基序列信息的目标区域。
(100pg以下的人类基因组dna)
本发明的判别来源的方法中,作为多重pcr的模板dna的人类基因组dna只要是100pg以下,则并无特别限定,优选为1pg以上且100pg以下,更优选为5pg以上且100pg以下,进一步优选为10pg以上且50pg以下。
对下限值进行说明。人类基因组dna约为30亿碱基对,这相当于约5pg。本发明中实施的多重pcr中,用150~200碱基对的各扩增子对人类基因组dna上的约650处同时进行扩增,由此能够分析约11万碱基对。这相当于人类基因组dna的0.004%,若换算为质量,则20fg成为理论上的检测下限值。但是实际上,若考虑dna聚合酶或引物稳定性、成为分析对象的人类基因组dna的状态,则优选为1pg以上,更优选为5pg以上,进一步优选为10pg以上。
(人类基因组dna上的区域)
本发明的判别来源的方法中,“人类基因组dna上的区域”是指存在具有基因多态性、单基因疾病、多因子疾病的可能性的基因区域的基因组dna上的区域。其中,区域的长度并无特别限定,1个碱基以上即可。选择目标区域的人类基因组上的区域可存在于基因区域及非基因区域中的任一区域。其中,基因区域包含基因多态性的区域。并且,非基因区域包含假基因、间隔物、响应元件及复制起点等非重复序列、以及串联重复序列及分散重复序列等重复序列。
基因多态性例如为snp(singlenucleotidepolymorphism:单核苷酸多态性)、snv(singlenucleotidevariant:单核苷酸变异)、strp(shorttandemrepeatpolymorphism:短串联重复序列多态性)、变异、以及插入和/或缺失(indel)等。
人类基因组dna上的区域的数量并无特别限定。这是因为,人类基因组dna上的区域为选择目标区域时的候选列表,即使有数量庞大的列表,也并非必需对其所有进行分析。
(目标区域)
目标区域为从上述人类基因组dna上的区域作为获取碱基序列信息的对象来选择的区域。其中,选择的目标并不限定于检测与各区域相关的基因多态性。并且,选择的目标并不限定于1个,可以具有2个以上的目标。
作为目标区域来选择的人类基因组dna上的区域的数量只要为至少1个,则并无特别限定,优选为3处以上,更优选为5处以上,进一步优选为10处以上。
<dna提取工序>
dna提取工序中,从人源试样提取人类基因组dna。
人源试样只要包含人类基因组dna,则并无特别限定,例如可举出细胞、毛发、血液及血迹等。人类基因组dna并不限定于存在于细胞内,也可以通过细胞被破坏等而逸出至细胞外。
(自细胞的dna提取)
自细胞的dna提取方法通过在蛋白质分解酶及表面活性剂的存在下加热来进行。通过该处理,细胞膜上的蛋白质及脂质成分溶解,能够将细胞内的基因组dna提取至细胞外。
进行dna提取的细胞优选为分离的细胞。分离细胞时,能够利用以往公知的细胞分离方法,如免疫染色、流式细胞仪、图像判别等。
<pcr扩增工序>
pcr扩增工序中,从在上述dna提取工序中获得的人类基因组dna,将100pg以下的人类基因组dna作为模板,通过聚合酶链反应对上述目标区域进行扩增。
关于用于聚合酶链反应的引物组合的设计方法,另外在“[用于聚合酶链反应的引物组合的设计方法]”中进行详细说明。
(聚合酶链反应)
聚合酶链反应使用dna聚合酶反复复制模板dna。在使其扩增的dna碱基序列的开始与结束的部分加以与模板dna进行杂交的较短的dna引物,并通过聚合酶开始复制。每次重复进行复制时,模板双链dna的2条链解离并分别被复制。
多重pcr能够使用通常的pcr中使用的耐热性dna聚合酶、反应缓冲液,各引物对与模板dna进行退火的温度不同,因此有时需要研究反应条件。因此,优选使用最适合于多重pcr的耐热性dna聚合酶、反应缓冲液,本发明中,更优选使用多重pcr检测试剂盒(takarabioinc.制造)进行反应。
关于引物组合的设计方法的详细内容将在后面进行叙述,因此在此叙述概要。
关于目标区域数量,从作为目标的染色体中特异性的dna序列区域,选择根据检查而所需的部位的对象区域,制作用于对各对象区域进行扩增的引物候选的碱基序列,并选拔引物碱基序列之间的互补性低的区域,由此对如100pg以下的微量的基因组dna,目标区域的扩增性也得到显著改善。
下一代测序仪技术虽然获得了飞跃性的进步,但用于序列分析的样品的制备需要非常复杂的工序。关于样品的预处理,在最广泛应用的基因组分析的情况下,在从样品提取核酸之后,需要(1)dna的片段化、(2)dna尺寸选择、(3)dna末端的平滑化处理、(4)向dna末端附加接头序列、(5)dna的纯化及(6)dna的扩增等工艺。
复杂的样品调整工序需要时间及劳力,必需确认工序是否适当进行。并且,各工序中逐渐产生偏差,在用作尤其以高水准要求结果的精度及准确度的医疗现场中的诊断工具时,需要减少该偏差。
为了解决这些问题,本发明中,利用使用后述的特定方法设计的引物来进行多重pcr,由此能够实现所选择的目标区域的更均匀的扩增。而且,通过磁珠对pcr产物进行纯化,由此能够进行更有效的扩增产物的回收。pcr反应溶液中残留有剩余引物、dntps(deoxynucleotides;脱氧核苷酸)及酶等掺杂物,因此有时会成为获得高精度的序列数据时的障碍。但是,通过利用磁珠对pcr扩增产物进行纯化,能够在显著抑制pcr扩增产物的损失的同时进行充分的纯化。此时,优选使用如下方法,即,从100g以下的非常少的dna,仅通过(1)dna的扩增及(2)dna的纯化这样的简单的样品调整工序,也均匀且高精度地扩增各种基因区域,可靠地检测细胞来源的信息。
被扩增的dna的定量例如能够利用测定260nm的波长的吸收度的超微量分光光度计nanodrop(thermofisherscientific制造)、利用激光荧光检测法的agilent2100bioanalyzer(agilenttechnologiesjapan,ltd.制造)或通过荧光法对双链dna进行定量的quantus荧光计(promegacorporation制造)等来进行。
《pcr扩增产物的纯化工序》
包含pcr扩增产物的反应液中混有酶、核苷酸、盐类及其他杂质,因此优选去除它们。作为核酸的纯化方法,例如,已知有利用苯酚/氯仿/乙醇的方法、在离液盐的存在下选择性地吸附于硅滤膜等硅载体的方法及利用在peg(polyethyleneglycol:聚乙二醇)存在下核酸选择性地与通过羧基修饰的磁珠结合的现象的方法等。
作为本发明的优选实施方式,多重pcr扩增产物能够选择利用磁珠的纯化方法。
(磁珠纯化工序)
对pcr扩增产物进行利用了作为顺磁微珠的磁珠的纯化,由此能够更有效地仅进行目标区域的分析。利用磁珠的纯化方法中,使核酸可逆性地结合于磁珠的粒子表面,用磁铁吸附磁珠,分离已扩增的dna片段与液体,由此能够有效地去除酶、dntps、pcr引物、引物二聚体及盐等掺杂物。利用磁珠的方法与其他纯化方法相比,样品损失较少且掺杂物的去除效率较高。作为其结果,dna序列工序中,能够有效地仅进行目标区域的分析。市售有磁珠,例如,能够使用ampurexp试剂盒(beckmancoulter,inc.制造)、nucleomag(takarabioinc.制造)或epinextdnapurificationhtsystem(epigentekgroupinc.制造)等。其中,优选利用ampurexp(beckmancoulter,inc.制造)进行纯化。
<dna测序工序>
解读在上述pcr扩增工序中获得的pcr扩增产物的dna碱基序列,获取上述目标区域的碱基序列信息。
pcr扩增产物的序列分析中优选利用下一代测序仪,尤其miseq(illumina,inc.制造)。利用下一代测序仪“miseq”对多个多重pcr产物进行测序时,需要对各多重pcr产物附加由6~8个碱基构成的样品识别序列(索引序列)以及用于与miseq流动池上的寡核苷酸序列杂交的p5序列及p7序列。通过这些序列的附加,能够一次测定最多96种多重pcr产物。作为对多重pcr产物的两个末端附加索引序列、p5序列及p7序列的方法,能够利用接头连接法(adapterligation)或pcr法等。
并且,混合多个多重pcr产物来通过miseq进行测定时,优选准确地定量各pcr产物。作为pcr产物的定量方法,还能够利用agilent2100bioanalyzer(agilenttechnologiesjapan,ltd.制造)或quantus荧光计(promegacorporation),但进一步优选通过定量pcr法测定多重pcr产物的方法。作为本发明中的定量方法,优选利用kapalibraryquantification试剂盒(nippongeneticsco.,ltd.制造)进行定量。
作为分析通过miseq获得的序列数据的方法,优选利用bwa(burrows-wheeleraligner;伯罗斯惠勒比对工具:li,h.、外1名、“fastandaccurateshortreadalignmentwithburrows-wheelertransform”、bioinformatics、2009年、第25卷、第14号、p.1754-1760.;li,h.、外1名、“fastandaccuratelong-readalignmentwithburrows-wheelertransform”、bioinformatics、2010年、第26卷、第5号、p.589-595),对已知的人类基因组序列映射,作为分析基因异常的方法,优选通过利用samtools(li,heng、外、“thesequencealignment/mapformatandsamtools”、bioinformatics、2009年、第25卷、第16号、p.2078-2079;sam来源于“sequencealignment/map”。)和/或bedtools(quinlan,a.r.、外1名、“bedtools:aflexiblesuiteofutilitiesforcomparinggenomicfeatures”、bioinformatics、2010年、第26卷、第6号、p.841-842),分析基因的变异或染色体数量的定量。
(序列读段数的确定)
dna测序工序中,优选还测定序列读段数。
例如,对于对对象区域进行pcr扩增来获得的dna片段,能够通过测序仪求出具有预先确定的140bp以上且180bp以下的区域的序列的扩增产物的扩增量(序列读段数)。作为基准(或者参考),通过测序仪求出鉴定为在犯罪搜查等中成为嫌疑对象的个人的细胞的细胞中,具有预先确定的140bp以上且180bp以下的区域的序列的扩增产物的扩增量(序列读段数)。
<来源判别工序>
根据上述碱基序列信息,判别上述人类基因组dna的来源。
dna的来源例如能够根据所获得的碱基序列信息的相似程度进行判别。原理上,从来源于同一人的dna可获得相同的碱基序列信息。但是,实际上,由于ado(alleledropout;等位基因脱扣)、dna的局部缺损或实验误差等,有时无法获得完全相同的碱基序列信息。
作为识别基因序列的个人差异的方法,通常通过检测等位基因中存在的基因多态性的方法进行。作为一例,父子辨别中,作为基因多态性的一种,还能够使用str。并且,作为识别个人差异的方法,还能够使用基因组碱基序列中的一个碱基变异并以1%以上的频率被观察的snps。本发明中,还能够利用下一代测序仪,进行dna序列状态的判定来确定从遗留品分离的细胞是否具有与嫌疑对象相同的str或snps。
[用于聚合酶链反应的引物组合的设计方法]
对本发明的获取来源于脊椎动物的单细胞的碱基序列信息的方法中实施的用于聚合酶链反应的引物组合的设计方法进行说明。
<用于聚合酶链反应的引物组合的设计方法的第1方式>
本发明中,用于聚合酶链反应的引物组合的设计方法的第1方式包含以下工序。另外,以下说明中,适当参考图2。
(a)对象区域选择工序,从至少1个目标区域选择对象区域(图2的s101)。
(b)引物候选碱基序列制作工序,根据上述脊椎动物的基因组dna上的上述对象区域的两端的各个附近区域的碱基序列,分别各制作至少1个用于对上述对象区域进行pcr扩增的引物候选的碱基序列(图2的s102)。
(c)局部比对工序,针对从在上述引物候选碱基序列制作工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分(图2的s103)。
(d)第1阶段选拔工序,根据上述局部比对评分,进行用于对上述对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔(图2的s104)。
(e)总体比对工序,针对从在上述第1阶段选拔工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分(图2的s105)。
(f)第2阶段选拔工序,根据上述总体比对评分,进行用于对上述对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔(图2的s106)。
(g)引物采用工序,采用在上述第1阶段选拔工序及上述第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述对象区域进行pcr扩增的引物的碱基序列(图2的s107)。
但是,上述(a)工序~上述(g)工序中,上述(c)工序及上述(d)工序这两个工序和上述(e)工序及上述(f)工序这两个工序可以为任意顺序或同时进行。即,可以在进行上述(c)工序及上述(d)工序之后进行上述(e)工序及上述(f)工序,也可以在进行上述(e)工序及上述(f)工序之后进行上述(c)工序及上述(d)工序,还可以同时进行上述(c)工序及上述(d)工序和上述(e)工序及上述(f)工序。
在进行上述(e)工序及上述(f)工序之后进行上述(c)工序及上述(d)工序时,上述(e)工序及上述(c)工序优选分别设为下述(e’)工序及下述(c’)工序。
(e’)总体比对工序,针对从在上述引物候选碱基序列制作工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
(c’)局部比对工序,针对从在上述第2阶段选拔工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分。
并且,同时进行上述(c)工序及上述(d)工序和上述(e)工序及上述(f)工序时,上述(e)工序优选设为下述(e’)工序。
(e’)总体比对工序,针对从在上述引物候选碱基序列制作工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
进一步设计引物时,若已制作引物候选的碱基序列,则从c)局部比对工序(图2的s103)设计引物,若未制作引物候选的碱基序列,则从b)引物候选碱基序列制作工序(图2的s102)或a)对象区域选择工序(图2的s101)设计引物(图2的s108、s109及s110)。
<用于聚合酶链反应的引物组合的设计方法的第2方式>
本发明中,作为至少1个目标区域包含2个以上的目标区域时的用于聚合酶链反应的引物组合的设计方法的派生方式之一的第2方式包含以下工序。另外,以下说明中,适当参考图3。
(a1)对象区域选择第1工序,从至少1个目标区域选择第1对象区域(图3的s201)。
(b1)引物候选碱基序列制作第1工序,根据上述脊椎动物的基因组dna上的上述第1对象区域的两端的各个附近区域的碱基序列,分别各制作至少1个用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列(图2的s202)。
(c1)局部比对第1工序,针对从在上述引物候选碱基序列制作第1工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分(图3的s203)。
(d1)第1阶段选拔第1工序,根据上述局部比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔(图3的s204)。
(e1)总体比对第1工序,针对从在上述第1阶段选拔第1工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分(图3的s205)。
(f1)第2阶段选拔第1工序,根据上述总体比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔(图3的s206)。
(g1)引物采用第1工序,采用在上述第1阶段选拔第1工序及上述第2阶段选拔第1工序二者中均被选拔的引物候选的碱基序列作为用于对上述第1对象区域进行pcr扩增的引物的碱基序列(图3的s207)。
但是,上述(a1)工序~上述(g1)工序中,上述(c1)工序及上述(d1)工序这两个工序和上述(e1)工序及上述(f1)工序这两个工序可以为任意顺序或同时进行。即,可以在进行上述(c1)工序及上述(d1)工序之后进行上述(e1)工序及上述(f1)工序,也可以在进行上述(e1)工序及上述(f1)工序之后进行上述(c1)工序及上述(d1)工序,还可以同时进行上述(c1)工序及上述(d1)工序和上述(e1)工序及上述(f1)工序。
在进行上述(e1)工序及上述(f1)工序之后进行上述(c1)工序及上述(d1)工序时,上述(e1)工序及上述(c1)工序分别优选设为下述(e1’)工序及下述(c1’)工序。
(e1’)总体比对第1工序,针对从在上述引物候选碱基序列制作第1工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
(c1’)局部比对第1工序,针对从在上述第2阶段选拔第1工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分。
并且,同时进行上述(c1)工序及上述(d1)工序和上述(e1)工序及上述(f1)工序时,上述(e1)工序优选设为下述(e1’)工序。
(e1’)总体比对第1工序,针对从在上述引物候选碱基序列制作第1工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
(a2)对象区域选择第2工序,从上述至少1个目标区域中还未被选择的目标区域选择第2对象区域(图3的s211)。
(b2)引物候选碱基序列制作第2工序,根据上述脊椎动物的基因组dna上的上述第2对象区域的两端的各个附近区域的碱基序列,分别各制作至少1个用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列(图3的s212)。
(c2)局部比对第2工序,针对从在上述引物候选碱基序列制作第2工序中制作的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分(图3的s213)。
(d2)第1阶段选拔第2工序,根据上述局部比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔(图3的s214)。
(e2)总体比对第2工序,针对从在上述第1阶段选拔第2工序中选拔的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分(图3的s215)。
(f2)第2阶段选拔第2工序,根据上述总体比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔(图3的s216)。
(g2)引物采用第2工序,采用在上述第1阶段选拔第2工序及上述第2阶段选拔第2工序二者中均被选拔的引物候选的碱基序列作为用于对上述第2对象区域进行pcr扩增的引物的碱基序列(图3的s217)。
但是,上述(a2)工序~上述(g2)工序中,上述(c2)工序及上述(d2)工序这两个工序和上述(e2)工序及上述(f2)工序这两个工序可以为任意顺序或同时进行。即,可以在进行上述(c2)工序及上述(d2)工序之后进行上述(e2)工序及上述(f2)工序,也可以在进行上述(e2)工序及上述(f2)工序之后进行上述(c2)工序及上述(d2)工序,还可以同时进行上述(c1)工序及上述(d1)工序和上述(e1)工序及上述(f1)工序。
在进行上述(e2)工序及上述(f2)工序之后进行上述(c2)工序及上述(d2)工序时,上述(e2)工序及上述(c2)工序分别优选设为下述(e2’)工序及下述(c2’)工序。
(e2’)总体比对第2工序,针对从在上述引物候选碱基序列制作第2工序中制作的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
(c2’)局部比对第2工序,针对从在上述第2阶段选拔第2工序中选拔的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分。
并且,同时进行上述(c2)工序及上述(d2)工序和上述(e2)工序及上述(f2)工序时,上述(e2)工序优选设为下述(e2’)工序。
(e2’)总体比对第2工序,针对从在上述引物候选碱基序列制作第2工序中制作的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
进一步设计引物时,反复进行a2)对象区域选择第2工序(图3的s211)至g2)引物采用第2工序(图3的s217)(图3的s208)。即,上述至少1个目标区域包含3个以上的目标区域的情况下,采用用于对还未从上述3个以上的目标区域选择的第3及其后的对象区域进行pcr扩增的引物的碱基序列时,针对上述第3及其后的对象区域,反复进行上述(a2)工序~上述(g2)工序的各工序。
<用于聚合酶链反应的引物组合的设计方法的第3方式>
本发明中,作为至少1个目标区域包含2个以上的目标区域时的用于聚合酶链反应的引物组合的设计方法的派生方式之一的第3方式包含以下工序。另外,以下说明中,适当参考图4。
(a-0)对象区域多个选择工序,从上述至少1个目标区域选择多个对象区域(图4的s301)。
(b-0)引物候选碱基序列多个制作工序,根据上述脊椎动物的基因组dna上的上述多个对象区域的各两端的各个附近区域的碱基序列,分别各制作至少1个用于分别对上述多个对象区域进行pcr扩增的引物候选的碱基序列(图4的s302)。
(c-1)第1局部比对工序,针对从在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列中用于对第1对象区域进行pcr扩增的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分(图4的s303)。
(d-1)第一第1阶段选拔工序,根据上述局部比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔(图4的s304)。
(e-1)第1总体比对工序,针对从在上述第一第1阶段选拔工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分(图4的s305)。
(f-1)第一第2阶段选拔工序,根据上述总体比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔(图4的s306)。
(g-1)第1引物采用工序,采用在上述第一第1阶段选拔工序及上述第一第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述第1对象区域进行pcr扩增的引物的碱基序列(图4的s307)。
但是,上述(c-1)工序~上述(g-1)工序中,上述(c-1)工序及上述(d-1)工序这两个工序和上述(e-1)工序及上述(f-1)工序这两个工序可以为任意顺序或同时进行。即,可以在进行上述(c-1)工序及上述(d-1)工序之后进行上述(e-1)工序及上述(f-1)工序,也可以在进行上述(e-1)工序及上述(f-1)工序之后进行上述(c-1)工序及上述(d-1)工序,还可以同时进行上述(c-1)工序及上述(d-1)工序和上述(e-1)工序及上述(f-1)工序。
在进行上述(e-1)工序及上述(f-1)工序之后进行上述(c-1)工序及上述(d-1)工序时,上述(e-1)工序及上述(c-1)工序分别优选设为下述(e’-1)工序及下述(c’-1)工序。
(e’-1)第1总体比对工序,针对从在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列中用于对第1对象区域进行pcr扩增的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
(c’-1)第1局部比对工序,针对在从上述第一第2阶段选拔工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分。
并且,同时进行上述(c-1)工序及上述(d-1)工序和上述(e-1)工序及上述(f-1)工序时,上述(e-1)工序优选设为下述(e’-1)工序。
(e’-1)第1总体比对工序,针对从在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列中用于对第1对象区域进行pcr扩增的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
(c-2)第2局部比对工序,针对从在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列中用于对第2对象区域进行pcr扩增的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分(图4的s313)。
(d-2)第二第1阶段选拔工序,根据上述局部比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔(图4的s314)。
(e-2)第2总体比对工序,针对从在上述第二第1阶段选拔工序中选拔的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分(图4的s315)。
(f-2)第二第2阶段选拔工序,根据上述总体比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔(图4的s316)。
(g-2)第2引物采用工序,采用上述第二第1阶段选拔工序及上述第二第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述第2对象区域进行pcr扩增的引物的碱基序列(图4的s317)。
但是,上述(c-2)工序~上述(g-2)工序中,上述(c-2)工序及上述(d-2)工序这两个工序和上述(e-2)工序及上述(f-2)工序这两个工序可以为任意顺序或同时进行。即,可以在进行上述(c-2)工序及上述(d-2)工序之后进行上述(e-2)工序及上述(f-2)工序,也可以在进行上述(e-2)工序及上述(f-2)工序之后进行上述(c-2)工序及上述(d-2)工序,还可以同时进行上述(c-1)工序及上述(d-1)工序和上述(e-1)工序及上述(f-1)工序。
在进行上述(e-2)工序及上述(f-2)工序之后进行上述(c-2)工序及上述(d-2)工序时,上述(e-2)工序及上述(c-2)工序分别优选设为下述(e’-2)工序及下述(c’-2)工序。
(e’-2)第2总体比对工序,针对从在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列中用于对第2对象区域进行pcr扩增的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
(c’-2)第2局部比对工序,针对从在上述第二第2阶段选拔工序中选拔的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分。
并且,同时进行上述(c-2)工序及上述(d-2)工序和上述(e-2)工序及上述(f-2)工序时,上述(e-2)工序优选设为下述(e’-2)工序。
(e’-2)第2总体比对工序,针对从在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列中用于对第2对象区域进行pcr扩增的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分。
进一步设计引物时,反复进行c-2)第2局部比对工序(图4的s313)至g-2)第2引物采用工序(图4的s317)(图3的s308)。即,上述至少1个目标区域包含3个以上的目标区域,在上述对象区域多个选择工序中选择3个以上的对象区域,且在上述引物候选碱基序列多个制作工序中制作用于分别对上述3个以上的对象区域进行pcr扩增的引物候选的碱基序列的情况下,采用用于对第3及其后的对象区域进行pcr扩增的引物的碱基序列时,针对上述第3及其后的对象区域,反复进行上述第2局部比对工序至上述第2引物采用工序的各工序。
<各工序的说明>
以下,对在本发明的获取来源于脊椎动物的单细胞的碱基序列信息的方法中实施的用于聚合酶链反应的引物组合的设计方法中包含的各工序进行详细说明。
《对象区域选择工序》
本说明书中,有时将对象区域选择工序s101(图2)、对象区域选择第1工序s201及对象区域选择第2工序s211(图3)以及对象区域多个选择工序s301(图4)简单地总称为“对象区域选择工序”。
(第1方式:对象区域选择工序s101)
图2中,示为“对象区域选择”(s101)。
第1方式中,(a)对象区域选择工序是从目标区域选择对象区域的工序。
(第2方式:对象区域选择第1工序s201及对象区域选择第2工序s211)
图3中,示为“对象区域选择第1”(s201)及“对象区域选择第2”(s211)。
第2方式中,(a1)对象区域选择第1工序是从目标区域选择第1对象区域的工序,(a2)对象区域选择第2工序是从还未被选择的目标区域选择第2对象区域的工序。
第2方式中,各选择1个目标区域。
(第3方式:对象区域多个选择工序s301)
图4中,示为“对象区域多个选择”(s301)。
第3方式中,(a-0)对象区域多个选择工序是从目标区域选择多个对象区域的工序。
第3方式中,选择多个目标区域。优选为选择所有目标区域作为对象区域。
《引物候选碱基序列制作工序》
本说明书中,有时将引物候选碱基序列制作工序s102(图2)、引物候选碱基序列制作第1工序s202及引物候选碱基序列制作第2工序s212(图3)以及引物候选碱基序列多个制作工序s302(图4)简单地总称为“引物候选碱基序列制作工序”。
(第1方式:引物候选碱基序列制作工序s102)
图2中,示为“引物候选碱基序列制作”(s102)。
第1方式中,(b)引物候选碱基序列制作工序是根据基因组dna上的上述对象区域的两端的各个附近区域的碱基序列,分别各制作至少1个用于对对象区域进行pcr扩增的引物候选的碱基序列的工序。
(第2方式:引物候选碱基序列制作第1工序s202及引物候选碱基序列制作第2工序s212)
图3中,示为“引物候选碱基序列制作第1”(s202)及“引物候选碱基序列制作第2”(s212)。
第2方式中,(b1)引物候选碱基序列制作第1工序是根据基因组dna上的上述第1对象区域的两端的各个附近区域的碱基序列,分别各制作至少1个用于对第1对象区域进行pcr扩增的引物候选的碱基序列的工序,(b2)引物候选碱基序列制作第2工序是根据基因组dna上的上述第2对象区域的两端的各个附近区域的碱基序列,分别各制作至少1个用于对第2对象区域进行pcr扩增的引物候选的碱基序列的工序。
第2方式中,针对1个对象区域进行引物候选的碱基序列的制作、引物候选的选拔及引物的采用,针对接下来的1个对象区域,反复进行相同的工序。
(第3方式:引物候选碱基序列多个制作工序s302)
图4中,示为“引物候选碱基序列多个制作”(s302)。
第3方式中,(b-0)引物候选碱基序列多个制作工序是根据基因组dna上的上述多个对象区域的各两端的各个附近区域的碱基序列,分别各制作至少1个用于分别对多个对象区域进行pcr扩增的引物候选的碱基序列的工序。
第3方式中,针对多个对象区域的所有区域制作引物候选的碱基序列,在之后的工序中反复进行选拔及采用。
(附近区域)
对象区域的两端的各个附近区域是指从对象区域的5’端靠外侧的区域及从对象区域的3’端靠外侧的区域的总称。对象区域的内侧不包含在附近区域。
附近区域的长度并无特别限定,但优选为能够通过pcr扩展的长度以下,更优选为希望扩增的dna片段长度的上限以下。尤其,优选为易实施集中选择和/或序列读段的长度。可根据pcr中使用的酶(dna聚合酶)的种类等适当变更。具体的附近区域的长度优选为20~500个碱基左右,更优选为20~300个碱基左右,进一步优选为20~200个碱基左右,更进一步优选为50~200个碱基左右。
(引物的设计参数)
并且,制作引物候选的碱基序列时,注意点与通常的引物的设计方法中的注意点相同,如引物的长度、gc含量(指所有核酸碱基中的鸟嘌呤(g)及胞嘧啶(c)的合计摩尔百分比。)、解链温度(指双链dna的50%解离而成为单链dna的温度。并且,有时来源于meltingtemperature而称为“tm值”。单位为“℃”。)及序列的偏移等。
·引物的长度
引物的长度(核苷酸数量)并无特别限定,但优选为15mer~45mer,更优选为20mer~45mer,进一步优选为20mer~30mer。若引物的长度在该范围内,则易设计特异性及扩增效率优异的引物。另外,“mer”为以核苷酸的个数表示多核苷酸的长度时的单位,1mer表示1个核苷酸。因此,例如15mer表示由15个核苷酸构成的多核苷酸。
·引物的gc含量
引物的gc含量并无特别限定,但优选为40摩尔%~60摩尔%,更优选为45摩尔%~55摩尔%。若gc含量在该范围内,则不易产生由高级结构引起的特异性及扩增效率的下降的问题。
·引物的tm值
引物的tm值并无特别限定,但优选在50℃~65℃的范围内,更优选在55℃~65℃的范围内。
引物的tm值的差在引物对及引物组合中,优选设为5℃以下,更优选设为3℃以下。
tm值能够利用oligoprimeranalysissoftware(molecularbiologyinsights公司制造)或primer3(http://www-genome.wi.mit.edu/ftp/distribution/software/)等软件计算。
并且,还能够由引物的碱基序列中的a、t、g及c的数量(分别设为na、nt、ng及nc),根据下述式通过计算来求出。
tm值(℃)=2(na+nt)+4(nc+ng)
tm值的计算方法并不限定于这些,能够通过以往公知的各种方法计算tm值。
·引物的碱基的偏移
引物候选的碱基序列优选设为整体上碱基无偏移的序列。例如,优选避免局部富gc的序列及局部富at的序列。
并且,优选还避免t和/或c的连续(聚嘧啶)以及a和/或g的连续(聚嘌呤)。
·引物的3’末端
而且,优选3’末端碱基序列避免富gc的序列或富at的序列。优选3’末端碱基为g或c,但并不限定于这些。
《特异性检查工序》
可根据在“引物候选碱基序列制作工序”中制作的各引物候选的碱基序列相对于染色体dna的序列互补性,实施评价引物候选的碱基序列的特异性的特异性检查工序。
特异性的检查中,进行染色体dna的碱基序列与引物候选的碱基序列的局部比对,局部比对评分小于预先设定的值时,能够评价为该引物候选的碱基序列相对于基因组dna的互补性低,特异性高。其中,优选对染色体dna的互补链也进行局部比对。这是因为引物为单链dna,而染色体dna为双链。并且,可代替引物候选的碱基序列,利用与此互补的碱基序列。
并且,也可将引物候选的碱基序列作为查询序列,对基因组dna碱基序列数据库进行同源性搜索。作为同源性搜索工具,例如可举出blast(basiclocalalignmentsearchtool:基本局部比对搜索工具)(altschul,s.a.、外4名、“basiclocalalignmentsearchtool”、journalofmolecularbiology、1990年、10月、第215卷、p.403-410)及fasta(pearson,w.r.、外1名、“improvedtoolsforbiologicalsequencecomparison”、美国科学院纪要、美国科学院、1988年、4月、第85卷、p.2444-2448)等。作为进行同源性搜索而得的结果,能够获得局部比对。
评分及局部比对评分的阈值均无特别限定,能够根据引物候选的碱基序列的长度和/或pcr条件等适当设定。使用同源性搜索工具时,可使用该同源性搜索工具的默认值。
例如,可考虑作为评分,采用匹配(互补碱基)=+1、不匹配(非互补碱基)=-1、indel(插入和/或缺失)=-3,将阈值设定为+15等。
引物候选的碱基序列与染色体dna上的预想外的位置的碱基序列具有互补性,且特异性低的情况下,利用该碱基序列的引物进行pcr时,有时对伪影进行扩增而非对象区域,因此排除。
《局部比对工序》
本说明书中,有时将局部比对工序s103(图2)、局部比对第1工序s203及局部比对第2工序s213(图3)以及第1局部比对工序s303及第2局部比对工序s313(图4)简单地总称为“局部比对工序”。
(第1方式:局部比对工序s103)
图2中,示为“局部比对”(s103)。
第1方式中,(c)局部比对工序是针对从在上述引物候选碱基序列制作工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分的工序。
(第2方式:局部比对第1工序s203及局部比对第2工序s213)
图3中,示为“局部比对第1”(s203)及“局部比对第2”(s213)。
第2方式中,(c1)局部比对第1工序是针对从在上述引物候选碱基序列制作第1工序中制作的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分的工序,(c2)局部比对第2工序是针对从在上述引物候选碱基序列制作第2工序中制作的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分的工序。
(第3方式:第1局部比对工序s303及第2局部比对工序s313)
图4中,示为“第1局部比对”(s303)及“第2局部比对”(s313)。
第3方式中,(c-1)第1局部比对工序是针对从选自在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列选择的用于对第1对象区域进行pcr扩增的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分的工序,(c-2)第2局部比对工序是针对从选自在上述引物候选碱基序列多个制作工序中制作的引物候选的碱基序列的用于对第2对象区域进行pcr扩增的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对上述组合中包含的2个碱基序列,在该比较的一部分序列包含上述2个碱基序列的3’末端的条件下,进行成对局部比对,从而求出局部比对评分的工序。
(局部比对的方法)
进行局部比对的碱基序列的组合可以是允许重复而选择的组合,也可以是不允许重复而选择的组合,但还未评价同一碱基序列的引物之间的引物二聚体形成性时,优选为允许重复而选择的组合。
关于组合的总数量,将进行局部比对的碱基序列的总数量设为p个,允许重复而选择时,为“ph2=p+1c2=(p+1)!/2(p-1)!”,不允许重复而选择时,为“pc2=p(p-1)/2”。
局部比对为对一部分序列进行的比对,能够局部性地检查互补性高的部分。
但是,本发明中,与通常对碱基序列进行的局部比对不同,在“所比较的一部分序列包含碱基序列的3’末端”这样的条件下,进行局部比对,以使所比较的一部分序列包含双方的碱基序列的3’末端。
而且,本发明中优选如下方式:在“所比较的一部分序列包含碱基序列的3’末端”这样的条件即“仅考虑所比较的一部分序列从其中一个序列的3’末端开始并在另一个序列的3’末端结束的比对”这样的条件下,进行局部比对,以使所比较的一部分序列包含双方的碱基序列的3’末端。
另外,局部比对可插入间隙。间隙表示碱基的插入和/或缺失(indel)。
并且,局部比对中,将在碱基序列对之间碱基为互补性的情况作为一致(匹配),将非互补性的情况作为不一致(不匹配)。
比对以对匹配、不匹配及插入和/或缺失分别赋予评分,并使合计评分成为最大的方式进行。评分适当设定即可。例如,可如下述表1那样设定评分系统。另外,表1中“-”表示间隙(插入和/或缺失(indel))。
[表1]
表1
例如,考虑对以下的表2所示的序列编号1及2的碱基序列进行局部比对。其中,评分如表1所示。
[表2]
表2
根据序列编号1及2的碱基序列制作表3所示的点阵(dotmatrix)。具体而言,将序列编号1的碱基序列从左至右以从5’至3’的方向排列,将序列编号2的碱基序列从下至上以从5’至3’的方向排列,对碱基为互补性的栅格记入“·”,由此获得表3所示的点阵。
[表3]
表3
根据表3所示的点阵获得以下的表4所示的一部分序列的比对(成对比对)(参考表3的斜线部分)。另外,表4中,以“|”表示匹配。
[表4]
表4
该(成对)比对中,有5处匹配,有4处不匹配,有1处插入和/或缺失(间隙)。
因此,基于该(成对)比对的局部比对评分成为(+1)×5+(-1)×4+(-1)×1=±0。
另外,比对(成对比对)并不仅限于在此例示的点阵法,能够通过动态规划法、词法或其他各种方法获得。
《第1阶段选拔工序》
本说明书中,有时将第1阶段选拔工序s104(图2)、第1阶段选拔第1工序s204及第1阶段选拔第2工序s214以及第一第1阶段选拔工序s304及第二第1阶段选拔工序s314简单地总称为“第1阶段选拔工序”。
(第1方式:第1阶段选拔工序s104)
图2中,示为“第1阶段选拔”(s104)。
第1方式中,(d)第1阶段选拔工序是根据上述局部比对评分,进行用于对上述对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔的工序。
(第2方式:第1阶段选拔第1工序s204及第1阶段选拔第2工序s214)
图3中,示为“第1阶段选拔第1”(s204)及“第1阶段选拔第2”(s214)。
第2方式中,(d1)第1阶段选拔第1工序是根据上述局部比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔的工序,(d2)第1阶段选拔第2工序是根据上述局部比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔的工序。
(第3方式:第一第1阶段选拔工序s304及第二第1阶段选拔工序s314)
图4中,示为“第一第1阶段选拔”(s304)及“第二第1阶段选拔”(s314)。
第3方式中,(d-1)第一第1阶段选拔工序是根据上述局部比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔的工序,(d-2)第二第1阶段选拔工序是根据上述局部比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第1阶段的选拔的工序。
(第1阶段选拔的方法)
预先设定局部比对评分的阈值(第1阈值)。
若局部比对评分小于第1阈值,则判断这2个碱基序列的对的二聚体形成性低,进行之后的工序。另一方面,若局部比对评分为第1阈值以上,则判断为这2个碱基序列的对的二聚体形成性高,对该对不进行之后的工序。第1阈值并无特别限定,能够适当设定。例如,可根据成为聚合酶链反应的模板的基因组dna的量等pcr条件设定第1阈值。
其中,上述“局部比对工序”中示出的例子中,考虑将第1阈值设定为“+3”的情况。
上面的例子中,局部比对评分为“±0”,小于作为第1阈值的“+3”,因此能够判断序列编号1及2的碱基序列的对的二聚体形成性低。
另外,对在局部比对工序s103、局部比对第1工序s203、局部比对第2工序s213、第1局部比对工序s303或第2局部比对工序s313中计算出局部比对评分的所有组合进行本工序。
《总体比对工序》
本说明书中,有时将总体比对工序s105(图2)、总体比对第1工序s205及总体比对第2工序s215(图3)以及第1总体比对工序s305及第2总体比对工序s315(图4)简单地总称为“总体比对工序”。
(第1方式:总体比对工序s105)
图2中,示为“总体比对”(s105)。
第1方式中,(e)总体比对工序是针对从在上述第1阶段选拔工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分的工序。
(第2方式:总体比对第1工序s205及总体比对第2工序s215)
图3中,示为“总体比对第1”(s205)及“总体比对第2”(s215)。
第2方式中,(e1)总体比对第1工序是针对从在上述第1阶段选拔第1工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分的工序,(e2)总体比对第2工序是针对从在上述第1阶段选拔第2工序中选拔的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分的工序。
(第3方式:第1总体比对工序s305及第2总体比对工序s315)
图4中,示为“第1总体比对”(s305)及“第2总体比对”(s315)。
第3方式中,(e-1)第1总体比对工序是针对从在上述第一第1阶段选拔工序中选拔的引物候选的碱基序列选择2个引物候选的碱基序列的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分的工序,(e-2)第2总体比对工序是针对从在上述第二第1阶段选拔工序中选拔的引物候选的碱基序列及已被采用的引物的碱基序列选择2个引物候选的碱基序列的组合和选择1个引物候选的碱基序列及1个已被采用的引物的碱基序列的组合的所有组合,对包含上述组合中包含的2个碱基序列的3’末端和预先设定的序列长度的碱基序列,进行成对总体比对,从而求出总体比对评分的工序。
(总体比对的方法)
关于总体比对评分,从由在“引物候选碱基序列制作工序”中制作的所有引物候选(已先进行“局部比对工序”及“第1阶段选拔工序”的情况下,存在局部比对评分小于第1阈值的引物候选的组合时,该组合中包含的所有引物候选。)及已采用的所有引物(仅限于存在已经采用的引物的情况。)构成的组提取2个引物,对包含3’末端和预先设定的序列长度的碱基序列,进行成对总体比对来求出。
进行总体比对的碱基序列的组合可以是允许重复而选择的组合,也可以是不允许重复而选择的组合,但还未评价同一碱基序列的引物之间的引物二聚体形成性时,优选为允许重复而选择的组合。
关于组合的总数量,将进行总体比对的碱基序列的总数量设为x个,允许重复而选择时,为“xh2=x+1c2=(x+1)!/2(x-1)!”,不允许重复而选择时,为“xc2=x(x-1)/2”。
总体比对为对“整个序列”进行的比对,能够检查整个序列的互补性。
但是,其中,“整个序列”为包含引物候选的碱基序列的3’末端和预先设定的序列长度的整个碱基序列。
另外,总体比对也可以插入间隙。间隙表示碱基的插入和/或缺失(indel)。
并且,总体比对中,将在碱基序列对之间碱基为互补性的情况作为一致(匹配),将非互补性的情况作为不一致(不匹配)。
比对以对匹配、不匹配及插入和/或缺失分别赋予评分,并使合计评分成为最大的方式进行。评分适当设定即可。例如,可如上述表1那样设定评分。另外,表1中“-”表示间隙(插入和/或缺失(indel))。
例如,考虑针对以下的表5所示的序列编号1及2的碱基序列,对各3’末端的3个碱基(大写字母部分。相当于“包含3’末端和预先设定的序列长度的碱基序列”。)进行总体比对。其中,评分系统如表1所示。
[表5]
表5
如以评分成为最大的方式,针对序列编号1的碱基序列的3’末端的3个碱基(大写字母部分)及序列编号2的3’末端的3个碱基(大写字母部分)的碱基序列进行总体比对,则能够获得以下的表6所示的比对(成对比对)。
[表6]
表6
该(成对)比对中,不匹配有3处,匹配及插入和/或缺失(间隙)均无。
因此,基于该(成对)比对的总体比对评分成为(+1)×0+(-1)×3+(-1)×0=-3。
另外,比对(成对比对)能够通过点阵法、动态规划法、词法或其他各种方法获得。
《第2阶段选拔工序》
本说明书中,有时将第2阶段选拔工序s106(图2)、第2阶段选拔第1工序s206及第2阶段选拔第2工序s216(图3)以及第一第2阶段选拔工序s306及第二第2阶段选拔工序s316(图4)简单地总称为“第2阶段选拔工序”。
(第1方式:第2阶段选拔工序s106)
图2中,示为“第2阶段选拔”(s106)。
第1方式中,(f)第2阶段选拔工序是根据上述总体比对评分,进行用于对上述对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔的工序。
(第2方式:第2阶段选拔第1工序s206及第2阶段选拔第2工序s216)
图3中,示为“第2阶段选拔第1”(s206)及“第2阶段选拔第2”(s216)。
第2方式中,(f1)第2阶段选拔第1工序是根据上述总体比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔的工序,(f2)第2阶段选拔第2工序是根据上述总体比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔的工序。
(第3方式:第一第2阶段选拔工序s306及第二第2阶段选拔工序s316)
图4中,示为“第一第2阶段选拔”(s306)及“第二第2阶段选拔”(s316)。
第3方式中,(f-1)第一第2阶段选拔工序是根据上述总体比对评分,进行用于对上述第1对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔的工序,(f-2)第二第2阶段选拔工序是根据上述总体比对评分,进行用于对上述第2对象区域进行pcr扩增的引物候选的碱基序列的第2阶段的选拔的工序。
(第2阶段选拔的方法)
预先设定总体比对评分的阈值(还称为“第2阈值”。)。
若总体比对评分小于第2阈值,则判断为这2个碱基序列的组合的二聚体形成性低,进行之后的工序。
另一方面,若总体比对评分为第2阈值以上,则判断为这2个碱基序列的组合的二聚体形成性高,对该组合不进行之后的工序。
第2阈值并无特别限定,能够适当设定。例如,可根据成为聚合酶链反应的模板的基因组dna的量等pcr条件设定第2阈值。
另外,能够通过将引物的3’末端至数个碱基的碱基序列设为同一碱基序列,使对包含各引物的碱基序列的3’末端和预先设定的碱基数的碱基序列,进行成对总体比对来求出的总体比对评分小于第2阈值。
在此,考虑在上述“总体比对工序”中示出的例子中将第2阈值设定为“+3”的情况。
上面的例子中,总体比对评分为“-3”,小于作为第2阈值的“+3”,因此能够判断为序列编号1及2的碱基序列的对的二聚体形成性低。
另外,对在总体比对工序s105、总体比对第1工序s205、总体比对第2工序s215、第1总体比对工序s305或第2总体比对工序s315中计算出总体比对评分的所有组合进行本工序。
并且,为了减少计算量,优选先实施“总体比对工序”及“第2阶段选拔工序”这两个工序,对通过“第2阶段选拔工序”的引物的碱基序列的组合实施“局部比对工序”及“第1阶段选拔工序”这两个工序。尤其,对象区域的数量越增加,引物候选的碱基序列数量越增加,减少计算量的效果越大,能够实现整个处理的快速化。
这是因为,在“总体比对工序”中对“预先设定的序列长度”这样的短长度的碱基序列进行总体比对,因此计算量少于在包含3’末端这样的条件下从整个碱基序列发现互补性高的一部分序列的局部比对评分,能够快速进行处理。另外,通常已知的算法中,已知若为针对相同长度的序列的比对,则总体比对的速度比局部比对快。
《扩增序列长度检查工序》
可实施扩增序列长度检查工序,其对在“第1阶段选拔工序”及“第2阶段选拔工序”中判断为引物二聚体形成性低的引物候选的碱基序列的组合,计算染色体dna上的引物候选的碱基序列的端部之间的距离,并判断该距离是否在预先设定的范围内。
若碱基序列的端部之间的距离在预先设定的范围内,则能够判断为该引物候选的碱基序列的组合能够对对象区域适当进行扩增的可能性高。引物候选的碱基序列的端部之间的距离并无特别限定,能够根据酶(dna聚合酶)的种类等pcr条件适当设定。例如,能够设定为100~200个碱基(对)的范围内、120~180个碱基(对)的范围内、140~180个碱基(对)的范围内、140~160个碱基(对)的范围内、160~180个碱基(对)的范围内等各种范围。
《引物采用工序》
本说明书中,有时将引物采用工序s107(图2)、引物采用第1工序s207及引物采用第2工序s217(图3)以及第1引物采用工序s307及第2引物采用工序s317(图4)简单地总称为“引物采用工序”。
(第1方式:引物采用工序s107)
图2中,示为“引物采用”(s107)。
第1方式中,(g)引物采用工序是采用在上述第1阶段选拔工序及上述第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述对象区域进行pcr扩增的引物的碱基序列的工序。
(第2方式:引物采用第1工序s207及引物采用第2工序s217)
图3中,示为“引物采用第1”(s207)及“引物采用第2”(s217)。
第2方式中,(g1)引物采用第1工序是采用在上述第1阶段选拔第1工序及上述第2阶段选拔第1工序二者中均被选拔的引物候选的碱基序列作为用于对上述第1对象区域进行pcr扩增的引物的碱基序列的工序,(g2)引物采用第2工序是采用上述第1阶段选拔第2工序及上述第2阶段选拔第2工序二者中均被选拔的引物候选的碱基序列作为用于对上述第2对象区域进行pcr扩增的引物的碱基序列的工序。
(第3方式:第1引物采用工序s307及第2引物采用工序s317)
图4中,示为“第1引物采用”(s307)及“第2引物采用”(s317)。
第3方式中,(g-1)第1引物采用工序是采用在上述第一第1阶段选拔工序及上述第一第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述第1对象区域进行pcr扩增的引物的碱基序列的工序,(g-2)第2引物采用工序是采用在上述第二第1阶段选拔工序及上述第二第2阶段选拔工序二者中均被选拔的引物候选的碱基序列作为用于对上述第2对象区域进行pcr扩增的引物的碱基序列的工序。
(引物采用的方法)
引物采用工序中,采用如下引物候选的碱基序列作为用于对对象区域进行扩增的引物的碱基序列,即,针对各引物候选的碱基序列,在所比较的一部分序列包含该碱基序列的3’末端这样的条件下,进行成对局部比对来求出的局部比对评分小于第1阈值,且针对包含各引物候选的碱基序列的3’末端和预先设定的碱基数的碱基序列,进行成对总体比对来求出的总体比对评分小于第2阈值。
例如,考虑采用表7所示的序列编号1及2的碱基序列作为用于对对象区域进行扩增的引物的碱基序列。
[表7]
表7
如已记载,局部比对评分为“±0”,因此小于作为第1阈值的“+3”,并且,总体比对评分为“-3”,因此小于作为第2阈值的“+3”。
因此,能够采用以序列编号1表示的引物候选的碱基序列及以序列编号2表示的引物候选的碱基序列作为用于对对象区域进行扩增的引物的碱基序列。
《其他目标区域的引物设计》
可在对1个目标区域采用引物之后,进一步对其他目标区域设计引物。
第1方式中,若在引物候选碱基序列制作工序s102中制作了针对欲设计引物的目标区域的引物候选的碱基序列,则从局部比对工序s103开始进行。若未制作针对下一目标区域的引物候选的碱基序列,则由于在对象区域选择工序s101中未选择下一目标区域,因此在对象区域选择工序s101中选择下一目标区域,并在引物候选碱基序列制作工序s102中制作针对该目标区域的引物候选的碱基序列之后,进行局部比对工序s103之后的工序。
第2方式中,从对象区域选择第2工序s211开始反复进行。
第3方式中,由于已在引物候选碱基序列多个制作工序s302中制作了针对在对象区域多个选择工序s301中选择的目标区域的引物候选的碱基序列,因此从第2局部比对工序s313开始反复进行。
《引物等的设计中的特征点》
总的来说,选择目标区域之后的引物等的设计中的特征点在于,选择多个特定的对象区域,搜索附近的碱基序列,确认与已提取的各个引物组合的互补性,选择互补性少的碱基序列,由此获得扩增对象中包含对象区域且不会相互互补的引物组。
引物的碱基序列的互补性确认中的特征点在于,以如下方式制作引物组,即,利用局部比对降低整个序列的互补性,且对引物的碱基序列的端部,利用总体比对降低互补性。
[识别个人的方法]
本发明的识别个人的方法利用上述的从100pg以下的人类基因组dna判别其来源的方法。
人类的基因中,作为因个人而不同的区域,已知有短重复单元的重复次数的str(shorttandemrepeat;串联重复序列)基因座的长度或具有1个碱基取代为其他碱基的信息的基因座即snp(singlenucleotidepolymorphisms:单核苷酸多态性),通过检查多个str基因座的长度或snp基因座的序列,能够识别个人。
例如,从灾害时的遗体的鉴定或犯罪搜查等中欲进行dna鉴定的对象的毛发或血迹等中包含的极微量的dna,也能够以高精度获取碱基序列信息。大多数情况下,欲进行个人识别的对象的个人遗留品等很少包含可通常获取的体细胞,还有作为细胞数可回收10个左右的情况。这种情况下,以往的dna扩增技术中,通常使用针对从10个左右以下的各细胞提取混合的量的dna,通过全基因组扩增法对成为分析对象的dna进行扩增,间接确定该扩增产物的碱基序列的方法,但该方法被指出如下问题,即,因所扩增的区域的不均匀化导致产生未被扩增的区域或因dna合成酶的扩增错误导致获得未反映准确的碱基序列信息的扩增产物,由此未必一定能够获取高精度的序列信息。而且,即使在不进行全基因组扩增而是直接从极微量dna进行pcr的情况下,也存在如下课题,即,会产生因模板dna量极少而产生的过度的扩增反应引起的不均匀化或为了获取碱基序列信息而使用的引物彼此的非特异性扩增(引物二聚体),无法获取准确的碱基序列信息。
成为灾害时或犯罪搜查中的分析对象的dna未必一定是完整形态的基因组dna,从细胞核露出并通过物理作用或以紫外线为代表的化学作用而片段化的情况也不少。在这种极微量的片段化的dna中,也能够获取基因序列信息。
本发明的识别个人的方法中,从在灾害时的遗体的鉴定或犯罪搜查等中欲进行dna鉴定的对象的毛发或血迹等中包含的极微量的dna,也能够以高精度获取碱基序列信息。大多数情况下,欲进行个人识别的对象的个人遗留品等很少包含可通常获取的体细胞,还有作为细胞数可回收10个左右的情况。这种情况下,通过本发明,也能够更加精度良好地进行个人识别。
[分析造血干细胞的植活程度的方法]
本发明的分析造血干细胞的植活程度的方法利用上述的从100pg以下的人类基因组dna判别其来源的方法。
称为嵌合性检查的检查是用作确认所移植的造血干细胞在很难生成正常的血液细胞的疾病的患者体内植活何种程度的指标的检查,这种检查中,也能够从少量的dna量精度良好地获取碱基序列信息。
作为造血器官的肿瘤或再生障碍性贫血等疾病的根治疗法而进行骨髓移植。此时,移植之后的健康提供者(供体)的骨髓细胞的植活和患者(受体)的细胞根除的确认对移植的成败判定、预后的判定而言极其重要。移植之后,有患者体内混合有来源于患者的细胞和来源于健康提供者的细胞的期间,称为混合嵌合状态。对该状态进行数值定量化的检查法为嵌合性检查。
嵌合性检查分为t淋巴细胞、t淋巴细胞以外的细胞分别进行分析。
当前,主要利用的嵌合性检查法中有str法,检查细胞的str。预先检查提供造血干细胞的供体的str和接受造血干细胞的移植的患者的str,事先确定长度互不相同的str基因座。移植肝细胞之后,若获取血液中的具有多个核的细胞,混合所提取的dna来进行观察,则在具有来源于供体的移植细胞的str的长度的信息最终成为100%的时点,能够明确地确认造血干细胞植活的状态。
检查snp时,通过下一代测序仪解读该snp基因座的snp序列所存在的数量,在供体的snp的信息成为100%的时点,能够明确地确认造血干细胞植活的状态。
利用snp的信息进行识别时,所比较的snp基因座的数量越多,识别的准确性越提高。对在个人之间具有相同的snp序列的概率(p)进行了计算,其结果,snp序列在1处成为p=5.2×10-1,在5处成为p=3.8×10-2,在10处成为p=1.45×10-3,在15处成为p=5.5×10-5,在20处成为p=2.09×10-6。即,若能够比较20处左右的snp序列,就能够以高精度识别造血干细胞的植活程度。
t淋巴细胞的检测方法具有:通过流式细胞仪法获取来源于试样液的细胞的信息,并根据所获取的信息,将目标细胞分取至排列有具有开口的孔的容器的工序;及拍摄分取至容器的细胞,并根据该图像确定t淋巴细胞候选细胞的工序。在利用流式细胞仪进行的分取之前进行染色。首先,准备包含目标细胞的分析对象试样。向分析对象试样例如混合溶血剂及用于免疫染色的被荧光标记的抗体,通过温育,对细胞进行免疫染色。通过对细胞进行免疫染色来制备试样液s。从光源向血液细胞照射激光束等。通过激光束的照射,基于血液细胞的免疫染色的荧光标记被激励,血液细胞通过基于免疫染色的荧光标记发出荧光。通过检测器检测该荧光强度。根据检测出的信息,在流动池被施加超声波的细胞带电为正或负。细胞通过偏转电极板时,通过带电的液滴靠近任一偏转电极板,基本上在容器的1个孔分取1个细胞。
以下,通过实施例对本发明进行更具体说明,但本发明并不限定于该实施例。
实施例
[实施例1]
1.分析dna的制备
(1)基因组dna
用dnase(dna分解酶)游离超纯水稀释从人类培养细胞提取混合的基因组dna,制备了基因组dna浓度为2.5ng/μl、250pg/μl、125pg/μl、25pg/μl、12.5pg/μl及2.5pg/μl的基因组dna水溶液。
将各浓度的基因组dna水溶液分别向0.2ml的pcr管各添加4μl。各pcr管中包含的基因组dna量分别为10ng、1ng、500pg、100pg、50pg及10pg。
(2)毛发dna
从男性志愿者(工作人员a)采集了1根毛发。
将所采集的毛发切分为毛根部和毛干,将其毛根部加入到0.2ml的pcr管中。
加入毛根部的pcr管中包含的基因组dna量约为50pg。
2.蛋白质溶解处理
在加入了4μl的分析dna的0.2ml的pcr管,添加了细胞溶解缓冲液(10mmol/l的tris-盐酸ph7.5,0.5mmol/l的乙二胺四乙酸,20mmol/l的氯化钾,0.007%(w/v)十二烷基硫酸钠,13.3μg/ml蛋白酶k)5μl。
将各pcr管在50℃下加热60分钟,通过蛋白酶k溶解了蛋白质。接着,在95℃下加热5分钟,使蛋白酶k失活。
在12℃下保存已进行蛋白质溶解处理的分析dna(以下,有时称为“蛋白质溶解物”。),直至下次使用。
3.分析dna的pcr扩增
(1)引物的制作
根据包含人类snps(singlenucleotidepolymorphisms;单核苷酸多态性)的647处的染色体位置或基因的碱基序列信息制作了多重pcr中使用的引物。
针对引物2001-f(序列编号1)及引物2001-r(序列编号2),在以下的表8示出局部比对,在表9示出总体比对。
评分设为匹配=+1、不匹配=-1及间隙(插入和/或缺失)=-1,对各位置的评分的总和进行比对、评分。
将局部比对评分的阈值设为“+3”,用于多重pcr的引物彼此的局部比对评分需小于该阈值。
并且,将总体比对评分的阈值设为“±0”,用于多重pcr的引物彼此的总体比对评分需小于该阈值。
[表8]
表8
局部比对评分为匹配(+1)×5+不匹配(-1)×4+插入和/或缺失(-1)×1=±0。
因此,局部比对评分小于阈值。
[表9]
表9
总体比对评分为匹配(+1)×0+不匹配(-1)×3+插入和/或缺失(-1)×0=-3。
因此,总体比对评分小于阈值。
针对所制作的引物的一部分,将各引物对的引物的名称、碱基序列及序列长度示于表10,将各引物对的扩增部位及扩增产物尺寸示于表11。
[表10]
表10
[表11]
表11
(2)基于单重pcr的扩增的确认
为了确认是否能够利用所制作的引物对所希望的扩增部位特异性地进行扩增,对各引物对进行了单重pcr。
a)反应液的制备
制备了以下的反应液。
另外,基因组dna为从人类培养细胞提取的基因组dna,multiplexpcrmix1及multiplexpcrmix2为takaramultiplexpcrassaykit(takarabioinc.制造)中包含的试剂。
b)单重pcr的反应条件
单重pcr的反应条件设为如下,即,初始热变性(94℃,60秒)之后,[热变性(94℃,30秒)-退火(60℃,90秒)-延伸(72℃,30秒)]的热循环×30循环。
c)pcr扩增产物的确认
将反应结束之后的反应液的一部分进行琼脂糖凝胶电泳,确认了pcr扩增产物的有无及尺寸。
关于647对引物对能够确认到基于单重pcr的特异性的扩增。
(3)基于多重pcr的扩增
a)反应液的制备
制备了以下反应液。
另外,模板dna为在上述“2.蛋白质溶解处理”中保存的“蛋白质溶解物”,引物混合物为以各引物的最终浓度成为50nmol/l的方式混合647对引物对的混合物,multiplexpcrmix1及multiplexpcrmix2为takaramultiplexpcrassaykit(takarabioinc.制造)中包含的试剂。
b)多重pcr的反应条件
多重pcr的反应条件设为如下,在初始热变性(94℃,60秒)之后,[热变性(94℃,30秒)-退火(56.7℃,600秒)-延伸(72℃,30秒)]的热循环×32循环。
c)pcr反应产物的纯化
利用agencourtampurexp(beckmancoulter,inc.制造),根据附带的手册(instructionsforuse),对通过多重pcr获得的pcr反应产物进行了纯化。
具体而言,向25μl的pcr反应液添加45μl的ampurexp试剂,充分混合,并在室温下静置5分钟,使pcr反应产物结合于磁珠。
接着,利用磁性表架(magnastand,nippongeneticsco.,ltd.制造)的磁力分离结合有pcr反应产物的磁珠,去除了掺杂物。
用70%(v/v)乙醇将结合有pcr反应产物的磁珠清洗2次之后,用40μl的tris-盐酸缓冲液溶出了结合于磁珠的dna。
4.dna测序
(1)序列库混合物的制备
为了利用下一代测序仪(miseq,illumina,inc.制造)进行双索引、测序,将包含流动池结合用序列(p5序列或p7序列)、样品识别用索引序列及测序引物结合用序列的2种接头分别附加于通过多重pcr获得的dna片段的两端。
具体而言,使用各1.25μm的表12所示的引物d501~d508及d701~d712,利用multiplexpcrassaykit进行pcr,由此进行了序列向两个末端的附加。pcr条件设为如下,即,在初始热变性(94℃,3分钟)之后,[热变性(94℃,45秒)-退火(50℃,60秒)-延伸(72℃,30秒)]的热循环×5循环,接着,[热变性(94℃,45秒)-退火(55℃,60秒)-延伸(72℃,30秒)]的热循环×11循环。
利用agencourtampurexp(beckmancoulter,inc.制造)对所获得的pcr产物进行纯化之后,利用kapalibraryquantificationkits(nippongeneticsco.,ltd.制造)定量了dna。
以索引序列不同的各样品的dna浓度分别成为1.5pm的方式进行混合来设为序列库混合物。
(2)扩增产物的序列分析
利用miseqreagentkitv2300cycle(illumina,inc.制造),对所制备的序列库混合物进行测序,获取了fastq文件。
利用bwa(burrows-wheeleraligner),从所获得的fastq数据对人类基因组序列(hg19)映射之后,通过samtools提取基因多态性信息,根据bedtools计算了各检测区域的序列读段数。
将序列读段的覆盖率为成为确定碱基序列的目标的19以上的比例示于图2。
在基因组dna的50pg~10ng的样品中,2次中的2次,覆盖率为19以上的比例均为0.95以上,超过了能够稳定地获取确定碱基序列所需的序列读段的0.8。
在基因组dna的10pg的样品中,2次中的1次,覆盖率为19以上的比例为0.9以上,超过了能够稳定地获取确定碱基序列所需的序列读段的0.8。
并且,在毛发dna的毛根样品中,2次中的2次,覆盖率为19以上的比例均为0.95以上,超过了能够稳定地获取确定碱基序列所需的序列读段的0.8。因此,能够在集中有来源于毛母细胞的dna的毛根部获取确定碱基序列所需的序列读段,确认到具有从1根毛发也能够确定碱基序列的检测灵敏度。
但是,在毛发dna的毛干样品中,2次中的2次,覆盖率为19以上的比例均约为0.2,未超过能够稳定地获取确定碱基序列所需的序列读段的0.8。这是因为毛干中实际上不包含核dna。
5.毛发的毛根部中包含的基因组dna的来源判别
已判明提供毛发的工作人员a为工作人员1~11中的任意1人,因此对从毛根中包含的基因组dna(50pg)获得的snps信息与工作人员1~11的snps信息进行比较,判别毛根中包含的基因组dna来源于工作人员1~11中的哪一工作人员。
工作人员a的snp信息根据从毛发的毛根部确定的碱基序列信息获取。
工作人员1~11的共11人的snps信息从预先构筑的工作人员snps数据库获取。
将工作人员9的snps图案作为参考,将工作人员a及工作人员1~11的snps信息[均质一致、异质不一致、ado(alleledropout;等位基因脱扣)、adi(alleledropin;等位基因插入)及均质不一致的个数]进行了视觉化。将其结果示于图3的累积纵条形图。
并且,将疑似同一人的似然度(概率)与疑似他人的似然度(概率)之比作为距离(似然度比)来进行了群分析。将其结果示于图4的系统树图。
通过以上,可知工作人员a的snps图案与工作人员9的snps图案一致,能够确认到为工作人员9。
- 还没有人留言评论。精彩留言会获得点赞!