丹参酮IIA噻吩吡啶类化合物及其制备方法和用途与流程
本发明属于医药技术领域,涉及一类具有心脑血管活性的丹参酮iia噻吩吡啶类化合物及其药学上可接受的盐和/或水合物。本发明还涉及这些化合物的制备方法,以及这些化合物在人类或其它哺乳动物的与血小板聚集相关的外周血管、内脏、肝脏和肾脏血管、心血管和脑血管的疾病或病症的治疗和/或预防中作为adp受体拮抗剂的用途。
背景技术:
心脑血管疾病,一直以来是致人类死亡的第一病因杀手,具有发病率高、致残率高、死亡率高、复发率高及并发症多的特点。目前,全球每年因心脑血管病死亡约1700万人,而我国每年死于心脑血管疾病近300万人,占我国每年总死亡病因的51%,已经成为我国乃至全球面临的重大公共卫生问题之一。近年来,随着基因组学、分子及细胞生物学的迅速发展,人们发现尽管心脑血管疾病有着复杂的致病机制,但可以明确的是,动脉粥样硬化斑块破裂或糜烂后的血栓形成是该类疾病发生和发展的决定性因素,而血小板聚集和凝血因子的级联启动则在血栓形成过程中扮演了关键角色(p.libby.mechanismsofacutecoronarysyndromesandtheirimplicationsfortherapy.n.engl.j.med.2013,368,2004-2013)。相应地,抗动脉粥样硬化、抗血小板聚集和抗凝一直以来是抗血栓心脑血管药物研究的重要方向。
然而,现有的抗血栓心脑血管药物要么以抗血小板聚集为主要机制(如阿司匹林、氯吡格雷、替罗非班等),要么以抗凝血为主要作用机制(如华法林、肝素、阿哌沙班、达吡加群酯等),却鲜有同时具有抗动脉粥样硬化、抗凝血和/或抗血小板聚集作用机制的药物。而在临床上,为了达到更好的血栓疾病治疗效果,常需将抗动脉粥样硬化药物、抗凝血药物和/或抗血小板聚集药物联合使用(郝素芳,夏云.注射用尤瑞克林与硫酸氢氯吡格雷联用治疗进展性脑梗死疗效观察[j].中国医药科学.2012,2,64-65;蓝昊.华法林联合氯吡格雷在冠心病合并心房颤动患者中的应用价值[j].心血管病防治知识.2017,5,61-62;姚均迪,沈彬等.阿托伐他汀对华法林抗凝作用的影响[j].海军医学杂志.2011,32,377-380。),但由于存在的药物-药物相互作用以及药物个体代谢的差异,这种联合用药往往也给患者带来了很大的安全隐患。因此,为了进一步满足血栓类疾病临床治疗药物的需求,开发具有多机制途径、综合治疗效果更优的抗血栓药物将具有重要研究和应用价值。
中药对复杂性疾病的治疗一直有着独特的优势,而以从传统活血化瘀中药中发现的天然活性成分为基础开发心脑血管药物,也成为我国现代新药创制的重要捷径。这其中,丹参酮iia及其磺酸钠类似物的发现就是重要的研究代表。丹参酮iia是从传统活血化瘀中药丹参中发现的脂溶性二萜醌类活性成分,约占药材含量的0.35%。研究发现,丹参酮iia具有广泛的药理作用如抗炎、抗动脉粥样硬化、扩张冠状动脉血管及调节免疫应答等,但由于脂溶性强和水溶性差的缺点,导致其口服生物利用极低,难以直接应用于临床。为了解决丹参酮iia溶解性差的问题,早期人们通过磺酸化反应得到了丹参酮iia磺酸钠,极大地提高了丹参酮iia的水溶性,并成功应用于临床冠心病、心绞痛和心肌梗死的辅助治疗。然而,磺酸基的存在虽然提高了丹参酮iia的水溶性,但也限制了其临床剂型的多样性,同时也导致其仅有的注射剂溶液ph偏低,刺激性大和化学稳定性差的安全性问题。此外,更重要的,丹参酮iia磺酸钠虽然在临床上得到了广泛应用,但其作用机制仍不够明确,作用靶点亦不清晰,从而严重影响了其临床用药的合理性和安全性,进而也导致了其目前作为心脑血管疾病辅助用药的临床定位。
近年来,人们对丹参酮iia也进行了大量的化学结构改造研究工作,并获得了多种结构类型的丹参酮iia类似物用于心脑血管疾病的预防和治疗。例如,①一种丹参酮iia磷酸酚酯衍生物及其制备方法,cn103382214a;②一种磺酰胺化合物的合成和应用,cn104341481a;③一种杂环磺酸衍生物的合成及其在药物治疗中的应用,cn104341482a;④一种磺酰胺衍生物的合成与药学应用,cn104341450a;⑤一种丹参酮iia衍生物及其制备和应用,cn104961794a;⑥一种丹参酮iia磷酸衍生物、及其合成和作为药物的应用,cn106478764a;⑦丹参酮iia衍生物在医药中的应用,cn105884856a;⑧丹参酮ⅱa丙烯酸或其钠盐及制备方法和应用,cn101974068a;⑨一种丹参酮类化合物17位酯化衍生物及其制备工艺和应用,cn106810593a;⑩一种丹参酮类化合物15位酰胺类衍生物及其制备工艺和应用,cn106831934a。然而,这些类似物虽然在水溶性和/或稳定性和/或生物活性上相比丹参酮iia有了一定程度的改善,但仍均未改变其靶点不清晰和机制不明确的关键问题。除此之外,该类化合物的制备路线较长,且得率较低,严重制约了它们的后续深入开发研究进展。因此,如何通过开展针对丹参酮iia的合理药物设计,发现作用机制明确,作用靶点清晰和生物利用度提高的新一代丹参酮类心脑血管药物引起了本发明人的关注。
adp即二磷酸腺苷,是由一分子腺苷与两个相连的磷酸根组成的核苷酸化合物,也是迄今为止人们发现最早和最重要的诱导血小板聚集的物质。人类血小板包括三种不同的adp受体:p2y1、p2y12、p2x1。p2x1是配体门控离子通道,p2y1、p2y12是与两种不同的g蛋白偶联的受体,其中在血小板上的数量上p2y12>p2y1,而p2y12也是现有临床adp受体阻滞剂氯吡格雷和普拉格雷及替格瑞洛主要作用靶点——通过选择性的抑制二磷酸腺苷(adp)与它的血小板受体p2y12的结合及继发的adp介导的糖蛋白gpiib/iiia复合物的活化,从而抑制血小板聚集。然而,现有adp阻滞剂虽然在临床上得到了广泛应用,但它们仍然存在很多疗效和安全方面的不足,例如氯吡格雷存在4~31%的氯吡格雷抵抗,严重影响了其疗效的发挥。又如,普拉格雷虽然具有更快更持久的抗血小板聚集作用,但出血的危险性相对氯吡格雷也有所增加(韩聪凡等.血栓性疾病抗血小板聚集药物研究进展.中国新药杂志.2016,12,1363-1369)。基于上述分析,结合中药在心脑血管等复杂性疾病的治疗优势,本发明人通过创造性发明,首次提供了一类丹参酮iia噻吩吡啶类化合物。在此基础上研究并明确了该类化合物对adp诱导的血小板聚集的抑制作用,显示出优良的用于心脑血管闭塞病的治疗前景。
技术实现要素:
本发明的目的是提供一类丹参酮iia噻吩吡啶化合物。
本发明的另一目的是提供用于预防和/或治疗心脑血管疾病的含有该类化合物药物组合物。
本发明的再一目的是提供该类化合物的制备方法。
本发明是关于一类丹参酮iia噻吩吡啶化合物,该类化合物在分子细胞水平上对adp的抑制作用及在体内动物水平上的治疗心脑血管疾病作用,具有预防和/或治疗心脑血管疾病的用途。
本发明提供一类丹参酮iia噻吩吡啶化合物及其可药用盐,这些化合物具有通式i所示的结构:
式中,r1为选自下组基团:h、甲酸c1~c6烷基酯基、c1~c6直链或支链烷基、c3~c6环烷基或环烯基、c1~c6直链或支链烷酰基、c3~c6环烷酰基、c2~c10直链或支链烯基、c2~c10直链或支链炔基、氰基、芳香基、芳烷基、杂环基、杂芳基。其中r1具体可为-h、-cooh、-cooch3、-cooch2ch3、-ch3、-ch2ch3、-ch(ch3)2、-ch2ch2ch3、-coch3、-coch2ch3、-cochch2ch2、-ph、-ph(2-cl)、-ph(2-f)、-ph(2-ome)、-ph(2-cf3)、并且r1优选为h、-cooch3。
r2选自以下任一结构基团:h、or3。其中r3为c1~c6烷酰基、c1~c6氧酰基、肉桂酰基、芳香酰基、杂芳香酰基;r3中所述肉桂酰基和芳香酰基中的芳基为苯基或者被1-4个选自卤素、羟基、硝基、氰基、三氟甲基、羧基、c1~c6烷基、c1~c6烷氧基中的基团所取代的苯基;r3中所述杂芳香酰基为具有1-3个选自o、n、s的杂原子的c5-c10杂芳香酰基。其中r2具体可为-coch3、-coch2ch3、-coch(ch3)2、-cochch2ch2、-cochchph、-cochchph(3,4-ome)、-coph、-coph(4-f)、-coph(4-ome)、-coph(3,4-ome)、-coph(4-no2)。
虚线为化学键或不存在。
本发明的部分优选的丹参酮iia噻吩吡啶类化合物如下所示。这些实施例举只对本发明做进一步说明,并不对本发明的范围构成任何限制。
其中众所周知任意上述列举的化合物的任意立构中心在未明示时可以是绝对(r)-或(s)-构型,也可以是其外消旋体混合物。
本发明还提供上述丹参酮iia噻吩吡啶类化合物可药用的盐。本发明可药用的盐可使用本领域熟知的标准程序获得,即由丹参酮iia噻吩吡啶类化合物与合适的酸反应得到的盐,如丹参酮iia噻吩吡啶化合物的盐酸盐、硫酸盐、磷酸盐、乙酸盐、柠檬酸盐、草酸盐、丙二酸盐、水杨酸盐、苹果酸盐、富马酸盐、琥珀酸盐、抗坏血酸盐、马来酸盐、酒石酸盐、枸橼酸盐、甲磺酸盐或羟乙磺酸盐。
根据本发明的第二方面,提供一种治疗心脑血管疾病的药物组合物,其中含有治疗有效量的至少一种选自式i的化合物和其可药用的盐化合物作为药剂。例如,作为可进行肠内或肠胃外施用的药物组合物的形式。
所述药物组合物可进一步包括可药用的载体、赋形剂、佐剂和/或辅料。所述载体、赋形剂、佐剂和/或辅料可以本领域任意技术人员熟知的方式(例如参见remington,thescienceandpracticeofpharmacy,21stedition(2005),part5,“pharmaceuticalmanufacturing”[publishedbylippincottwilliams&wilkins])进行生产,其包括将所述的式i的化合物及其药学可接受的盐,可选地联合其它有治疗价值的物质,连同适用的、无毒的、惰性的、治疗相容性固体或液体载体材料以及根据需要选用的常用药物佐剂制成盖仑制剂形式。
根据本发明的一种实施方式,本发明的药物组合物,可进一步包括治疗有效量的其他可药用的治疗剂作为活性成分,组成复方制剂。
根据本发明的第三方面,提供上述丹参酮iia噻吩吡啶类化合物或其可药用的盐的制备方法,该方法包括下列方法制备。
式i化合物的制备
以下缩写应用于整个说明书和实施例中:
acoh乙酸
adp腺苷二磷酸
bsa牛血清白蛋白
dmso二甲基亚砜
dmfn,n-二甲基甲酰胺
dpm每分钟衰变
lc-ms液相色谱-质谱
ph-苯基
rt室温
sds十二烷基硫酸钠
tlc薄层层析
v溶液体积
本发明式i化合物可按照如下通用方法制备:
本发明的r2取代基为h或o时,可按上式方法a)所示方法一步反应直接得到结构式i-a的目标化合物。丹参酮iia与取代醛和噻吩吡啶在lewis酸催化下发生mannich反应,得到丹参酮iia噻吩吡啶化合物(i-a)。
r2取代基为or3时,可按上式方法b)所示方法经三步反应得到结构式i-b的目标化合物。以三苯甲基保护的吡啶噻吩酮为起始原料,在碱作用下与r3x发生酰化得到中间体-1,然后经酸选择性水解得到关键中间体-2,最后再与丹参酮iia和取代醛在lewis酸催化下发生mannich反应,得到丹参酮iia噻吩吡啶化合物(i-b)。
优选地,在根据本发明的方法中,方法a)和方法b)中mannich反应步骤中所用溶剂可以是但不限于c1-c4醇、氯仿、二氯甲烷、乙酸乙酯、乙酸甲酯、n,n-二甲基甲酰胺、二甲基亚砜、乙醚、甲基叔丁基醚、四氢呋喃、乙腈、氯苯、二甲苯、甲苯、甲酸、乙酸或它们的两两混合溶液等;反应温度一般在rt~120oc,反应温度取决于具体化合物的反应活性情况。
优选地,在根据本发明的方法中,方法b)步骤1中酰化所用的碱类反应试剂可以是但不限于氢化钠、三乙胺、二异丙基乙二胺,并且酰化剂可为酰氯或酸酐。
优选地,在根据本发明的方法中,方法b)步骤2中水解所用的酸类反应试剂可以是但不限于盐酸或对甲苯磺酸。
优选地,在根据本发明的方法中,方法a)和b)步骤中mannich反应所用lewis酸可以为质子酸,也可以为金属lewis酸;优选为金属lewis酸如过渡金属和镧系金属盐。
优选地,在根据本发明的方法中,方法c)成盐步骤中所用溶剂可为水、c1-c4醇、二甲基亚砜、二甲基甲酰胺、丙酮、乙腈、氯仿、二氯甲烷、甲基叔丁基醚或它们的两两混合溶液,反应温度为rt~60oc;所用的酸ha可为盐酸、硫酸、磷酸、乙酸、柠檬酸、草酸、丙二酸、水杨酸、苹果酸、富马酸、琥珀酸、抗坏血酸、马来酸、酒石酸、枸橼酸、甲磺酸或羟乙磺酸;n表示结合酸的个数,n=0~2,具体可为0.5,1,1.5,2。
具体地,根据本发明的方法,以丹参酮iia为底物,与取代醛和衍生化的噻吩吡啶在lewis酸催化下经mannich反应直接得到式i-a和式i-b的系列丹参酮iia噻吩吡啶类化合物。通常用tlc和lc-ms来检测反应完成程度,反应完毕后一般用甲基叔丁基醚、乙酸乙酯或二氯甲烷等溶剂萃取,依次用饱和碳酸氢钠、水和饱和食盐水洗,经无水硫酸钠或者硫酸镁干燥,低温减压下除去溶剂。中间产物及最终产物用核磁共振及质谱检测确证。
根据本发明的第四方面,式i的化合物是adp受体拮抗剂。相应地,它们可用于治疗(包括联合治疗)心脑血管闭塞病,其中它们被广泛地用作血小板激活、聚集和脱粒的抑制剂,用作血小板解聚的促进剂或作为抗血栓剂。即本发明还提供上述丹参酮iia噻吩吡啶类化合物或其可药用的盐在制备预防或治疗血小板聚集引起的心脑血管疾病的药物中的应用。所述心脑血管疾病为冠心病、心肌梗塞、心绞疼、血栓溶解疗法或经皮冠状动脉腔内成形术有关的急性血管闭合、短暂性缺血发作、中风、间歇性跛行或冠状或外周动脉的旁路移植术、血管腔变窄、冠状动脉或静脉血管成形术后的再狭窄、长期血液透析患者血管通路通畅的维持、和肺血栓栓塞。
有益效果
本发明设计并合成了一类新型丹参酮iia噻吩吡啶结构类化合物,其对adp诱导的血小板聚集有明显的抑制作用。该类化合物不仅化学稳定性好,而且水溶性大大改善,显示出优异的成药性特征,可用于制备治疗心脑血管疾病药物。本发明化合物合成简单,易于制备,且合成原料丰富,具有显著的产业化开发研究价值。
附图说明
图1是本发明丹参酮iia噻吩吡啶类化合物体外抗adp诱导的血小板聚集活性实验结果。
图2本发明丹参酮iia噻吩吡啶类化合物抑制大鼠动静脉旁路血栓形成作用实验结果。
下面结合具体实施例对本发明作进一步阐释,但不限制本发明。本发明的实验操作具有通用性,不限于以下实施例中提到的具体化合物。
下述制备例中,1h-nmr用varianmercuryamx300型仪测定。ms用vgzab-hs或vg-7070型以及esquire3000plus-01005测定。所有反应溶剂在使用前均经过重新蒸馏,所使用的无水溶剂均是按照标准方法干燥处理获得。除说明外,所有反应均是在氩气保护下进行并用tlc跟踪,后处理时均经饱和食盐水和无水硫酸钠干燥过程。产品的纯化除说明外均使用硅胶(200-300目)的柱色谱纯化。
路线1
实施例1
将多聚甲醛(30mg,1mmol)、醋酸铜(2mg,0.01mmol)和盐酸噻吩吡啶(52mg,0.3mmol)加入到含有丹参酮iia(59mg,0.2mmol)二甲基亚砜和冰醋酸的混合溶液中(v/v=2:1,2ml),并于60oc下反应至完全。反应完成后,过滤,所得滤液减压蒸干后经硅胶柱色谱纯化(v石油醚:v乙酸乙酯=10:1~2:1),得到实施例1化合物jnld-1(68mg,收率76%)。ms-esi[m+h]+446.1;1hnmr(300mhz,cdcl3)δ7.65–7.52(m,2h),7.09(d,j=5.1hz,1h),6.74(d,j=5.1hz,1h),3.79(s,2h),3.67(s,2h),3.18(t,j=6.4hz,2h),2.98–2.88(m,4h),2.29(s,3h),1.84–1.74(m,2h),1.66–1.61(m,2h),1.30(s,6h)。
实施例2至实施例7的制备参考实施例1的操作,通过路线1得到,其中多聚甲醛可被其他取代醛如乙醛酸甲酯、乙醛酸乙酯、乙醛环丙酮和芳香醛取代,所得结果如下:
路线2
实施例6
制备步骤1:实施例6-1合成
在冰浴搅拌的条件下,向含有sm-1(397mg,1mmol)和三乙胺(201mg,2mmol)的乙腈溶液(2ml)中慢慢加入乙酸酐(204mg,2mmol),然后缓慢升至室温并继续反应2~3h至完全。tlc检测反应完成后,加水5ml淬灭,然后加入乙酸乙酯(10ml)萃取,有机相依次用饱和碳酸氢钠、食盐水洗涤,无水硫酸钠干燥后经减压浓缩至干得到白色固体,然后经乙醇重结晶得到实施例6-1化合物jnld-2(408mg,93%yield)。ms-esi[m+h]+440.1。
制备步骤2:实施例6-2合成
冰浴搅拌条件下,向含有实施例6-1化合物(219mg,0.5mmol)的丙酮溶液(2ml)中加入2.5mhcl丙酮溶液(2ml,5mmol)。然后升至rt~45oc并继续反应2~3h至完全。反应完成后,将析出沉淀固体过滤,丙酮洗涤(5ml),然后经减压干燥得到实施例6-2化合物(87mg,75%yield)。ms-esi[m+h]+198.0;1hnmr(300mhz,dmso-d6)δ9.76(brs,1h),6.54(s,1h),4.37−3.93(m,2h),3.42−3.28(m,2h),3.05(t,j=6.0hz,2h),2.26(s,3h)。
制备步骤3:实施例6合成
将多聚甲醛(30mg,1mmol)、醋酸锌(2mg,0.01mmol)和实施例6-1化合物(70mg,0.3mmol)加入到含有丹参酮iia(59mg,0.2mmol)二甲基亚砜溶液(2ml)中,并于60oc下反应24h。反应完成后,过滤,所得滤液减压蒸干后经硅胶柱色谱纯化(v石油醚:v乙酸乙酯=10:1~2:1),得到实施例6化合物jnld-2(90mg,收率89%)。ms-esi[m+h]+504.1;1hnmr(300mhz,cdcl3)δ7.66–7.52(m,2h),6.27(s,1h),3.80–3.65(m,4h),3.17(t,j=6.4hz,2h),2.98–2.89(m,4h),2.28(s,3h),2.25(s,3h),1.82–1.73(m,2h),1.67–1.62(m,2h),1.30(s,6h)。
实施例7
照实施例6中的制备方法,通过路线2,其中制备步骤1中以丙酸酐替代乙酸酐为原料,得实施例7化合物jnld-3(89mg,收率86%)。ms-esi[m+h]+518.3;1hnmr(300mhz,cdcl3)δ7.65–7.52(m,2h),6.26(s,1h),3.81–3.65(m,2h),3.16(t,j=6.4hz,2h),2.98–2.88(m,4h),2.55(q,2h,j=7.4hz),2.28(s,3h),1.82–1.75(m,2h),1.66–1.60(m,2h),1.29(s,6h),1.23(t,3h,j=7.4hz)。
实施例8
照实施例6中的制备方法,通过路线2,其中制备步骤1中以异丁酰氯替代乙酸酐为原料,得实施例8化合物jnld-4(84mg,收率79%)。ms-esi[m+h]+532.3;1hnmr(300mhz,cdcl3)δ7.66–7.52(m,2h),6.26(s,1h),3.80–3.65(m,4h),3.17(t,j=6.4hz,2h),2.97–2.88(m,4h),2.70–2.63(m,1h),2.28(s,3h),1.82–1.74(m,2h),1.66–1.61(m,2h),1.30(s,6h),1.27(d,j=2.1hz,3h),1.24(d,j=2.1hz,3h)。
实施例9
照实施例6中的制备方法,通过路线2,其中制备步骤1中以苯甲酰氯替代乙酸酐为原料,得实施例9化合物jnld-5(104mg,收率92%)。ms-esi[m+h]+566.3;1hnmr(300mhz,cdcl3)δ8.17(d,j=8.1hz,2h),7.72–7.52(m,5h),6.42(s,1h),3.79–3.64(m,4h),3.17(t,j=6.3hz,2h),2.97–2.89(m,4h),2.27(s,3h),1.82–1.75(m,2h),1.65–1.60(m,2h),1.29(s,6h)。
实施例10
照实施例6中的制备方法,通过路线2,其中制备步骤1中以3,4-二甲氧基苯甲酰氯替代乙酸酐为原料,得实施例10化合物jnld-6(109mg,收率87%)。ms-esi[m+h]+626.3;1hnmr(300mhz,cdcl3)δ7.86(dd,j=8.4,1.7hz,1h),7.70(d,j=1.7hz,1h),7.66–7.53(m,2h),6.95(d,j=8.4hz,1h),6.39(s,1h),3.85(s,6h),3.81–3.65(m,4h),3.17(t,j=6.4hz,2h),2.97–2.88(m,4h),2.28(s,3h),1.82–1.74(m,2h),1.66–1.60(m,2h),1.30(s,6h)。
实施例11
照实施例6中的制备方法,通过路线2,其中制备步骤1中以氯甲酸甲酯替代乙酸酐为原料,得实施例11化合物jnld-7(89mg,收率86%)。ms-esi[m+h]+520.3;1hnmr(300mhz,cdcl3)δ7.66–7.52(m,2h),6.40(s,1h),3.90(s,3h),3.80–3.64(m,4h),3.18(t,j=6.4hz,2h),2.98–2.88(m,4h),2.28(s,3h),1.81–1.59(m,4h),1.30(s,6h)。
实施例12
照实施例6中的制备方法,通过路线2,其中制备步骤1中以4-氟苯甲酰氯替代乙酸酐为原料,得实施例12化合物jnld-8(94mg,收率81%)。ms-esi[m+h]+584.3;1hnmr(300mhz,cdcl3)δ8.18(d,j=8.4hz,2h),7.67–7.58(m,4h),6.42(s,1h),3.79–3.66(m,4h),3.18(t,j=6.4hz,2h),2.99–2.89(m,4h),2.28(s,3h),1.82–1.75(m,2h),1.66–1.60(m,2h),1.30(s,6h)。
实施例13
照实施例6中的制备方法,通过路线2,其中制备步骤1中以肉桂酰氯替代乙酸酐为原料,得实施例13化合物jnld-9(76mg,收率64%)。ms-esi[m+h]+592.1;1hnmr(300mhz,cdcl3)δ7.85(d,j=15.9hz,1h),7.71–7.33(m,7h),6.56(d,j=15.9hz,1h),6.40(s,1h),3.81–3.64(m,4h),3.18(t,j=6.3hz,2h),2.97–2.89(m,4h),2.28(s,3h),1.81–1.73(m,2h),1.66–1.60(m,2h),1.30(s,6h)。
实施例14
照实施例6中的制备方法,通过路线2,其中制备步骤1中以烟酰氯替代乙酸酐为原料,得实施例14化合物jnld-10(88mg,收率78%)。ms-esi[m+h]+567.1;1hnmr(300mhz,cdcl3)δ9.34(m,1h),8.84−8.83(m,1h),8.41−8.38(m,1h),7.69–7.53(m,3h),6.42(s,1h),3.82–3.66(m,4h),3.17(t,j=6.3hz,2h),2.97–2.87(m,4h),2.28(s,3h),1.82–1.74(m,2h),1.66–1.60(m,2h),1.29(s,6h)。
实施例15
照实施例6中的制备方法,通过路线2,其中制备步骤3以三聚乙醛替代多聚甲醛,得实施例15化合物jnld-11(12mg,收率11%)。ms-esi[m+h]+518.3;1hnmr(300mhz,cdcl3)δ7.66–7.52(m,2h),6.27(s,1h),4.72–4.66(m,1h),3.68(s,2h),3.17(t,j=6.4hz,2h),2.97–2.88(m,4h),2.28(s,3h),2.26(s,3h),1.82–1.61(m,7h),1.31(s,3h),1.30(s,3h)。
实施例16
照实施例6中的制备方法,通过路线2,其中制备步骤3以丙醛替代多聚甲醛,得实施例16化合物jnld-12(10mg,收率9%)。ms-esi[m+h]+532.3;1hnmr(300mhz,cdcl3)δ7.65–7.53(m,2h),6.27(s,1h),4.53–4.37(m,1h),3.67(s,2h),3.17(t,j=6.3hz,2h),2.98–2.89(m,4h),2.28(s,3h),2.25(s,3h),1.82–1.74(m,6h),1.30(s,3h),1.29(s,3h),1.05–0.96(m,3h)。
实施例17
照实施例6中的制备方法,通过路线2,其中制备步骤3以丙酮醛替代多聚甲醛,得实施例17化合物jnld-13(79mg,收率73%)。ms-esi[m+h]+546.3;1hnmr(300mhz,cdcl3)δ7.66–7.52(m,2h),6.27–5.39(m,2h),3.70(s,2h),3.19(t,j=6.3hz,2h),2.99–2.89(m,4h),2.28(s,3h),2.25(s,3h),2.06(s,3h),1.82–1.61(m,4h),1.30(s,3h),1.29(s,3h)。
实施例18
照实施例6中的制备方法,通过路线2,其中制备步骤3中以2-环丙基丙酮醛替代多聚甲醛,得实施例18化合物jnld-14(78mg,收率68%)。ms-esi[m+h]+572.3;1hnmr(300mhz,cdcl3)δ7.66–7.52(m,2h),6.27–5.42(m,2h),3.70(s,2h),3.19(t,j=6.3hz,2h),2.99–2.87(m,4h),2.37–2.35(m,1h),2.28(s,3h),2.26(s,3h),1.84–1.73(m,2h),1.67–1.60(m,2h),1.31(s,3h),1.30(s,3h),1.15–1.09(m,2h),1.00–0.92(m,2h)。
实施例19
照实施例6中的制备方法,通过路线2,其中制备步骤3以乙醛酸甲酯替代多聚甲醛,得实施例19化合物jnld-15(93mg,收率83%)。ms-esi[m+h]+562.2;1hnmr(300mhz,cdcl3)δ7.66–7.52(m,2h),6.27–5.41(m,2h),3.73(s,3h),3.67(s,2h),3.19(t,j=6.3hz,2h),2.98–2.88(m,4h),2.28(s,3h),2.26(s,3h),1.82–1.73(m,2h),1.67–1.61(m,2h),1.30(s,3h),1.29(s,3h)。
路线3
实施例20
在氮气保护下,将化合物jnld-14(57mg,0.1mmol)溶于1ml乙醇溶液,然后加入0.5ml10%hcl水溶液并于室温条件下搅拌2天至反应完全。反应完成后,加入乙酸乙酯(5ml)和饱和食盐水(3ml)萃取,所得有机相减压蒸干后经硅胶柱色谱纯化(v石油醚:v乙酸乙酯=6:1~1:1),得到实施例20化合物jnld-20(41mg,收率78%)。ms-esi[m+h]+530.1;1hnmr(300mhz,cdcl3)δ7.66–7.53(m,2h),6.03(s,1h),5.51(s,1h),4.16–3.93(m3h),3.25–2.31(m,6h),2.28(s,3h),1.93–1.62(m,5h),1.31(s,3h),1.30(s,3h),1.15–1.09(m,2h),1.00–0.92(m,2h)。
实施例21
照实施例20中的制备方法,通过路线3,以jnld-15为原料,得实施例21化合物jnld-21(39mg,收率75%)。ms-esi[m+h]+520.2;1hnmr(300mhz,cdcl3)δ7.67–7.52(m,2h),6.04(s,1h),5.42(s,1h),4.18–3.92(m3h),3.75(s,3h),3.25–2.38(m,6h),2.29(s,3h),1.93–1.63(m,4h),1.32(s,3h),1.31(s,3h)。
实施例22
照实施例20中的制备方法,通过路线3,以jnld-2为原料,得实施例22化合物jnld-22(39mg,收率75%)。ms-esi[m+h]+462.1;1hnmr(300mhz,cdcl3)δ7.65–7.53(m,2h),6.05(s,1h),4.17–3.92(m,2h),3.80(s,2h),3.65–2.32(m,6h),2.28(s,3h),1.93–1.61(m,5h),1.30(s,6h)。
实施例23化合物jnld-15硫酸盐的制备
将溶有实施例19化合物jnld-15(56mg,0.1mmol)的乙醇溶液,于室温下缓慢加入12ml0.005mh2so4溶液,并搅拌至溶解;然后将所得红色溶液减压浓缩至3ml,放置室温下冷却析晶,过滤,冰水洗涤,得到jnld-15硫酸盐红色固体(42mg,收率75%)。ms-esi[m+h]+562.2;1hnmr(300mhz,cd3od)δ7.86–7.65(m,2h),6.96–5.68(m,2h),4.93–4.68(m,5h),3.80–3.17(m,6h),2.42(s,3h),2.38(s,3h),1.88–1.62(m,4h),1.35(s,3h),1.32(s,3h)。
实施例24化合物jnld-1盐酸盐的制备
将溶有实施例1化合物jnld-1(45mg,0.1mmol)的乙醇溶液,于室温下缓慢加入22ml0.005mhcl溶液,并搅拌至溶解;然后将所得红色溶液减压浓缩至2ml,放置室温下冷却析晶,过滤,冰水洗涤,得到jnld-1盐酸盐红色固体(41mg,收率69%)。ms-esi[m+h]+446.1;1hnmr(300mhz,cd3od)δ7.81–7.67(m,2h),7.45–7.40(m,1h),6.94(d,j=5.2hz,1h),4.85–4.60(m,4h),3.68–3.14(m,6h),2.39(s,3h),1.84–1.66(m,4h),1.35(s,3h),1.33(s,3h)。
实施例25水溶性研究
脂溶性强水溶性差是丹参酮iia成药性差的主要原因之一。为了明确本发明所得丹参酮iia噻吩吡啶类化合物的水溶性的改善情况,本实验初步研究了具有代表性的丹参酮iia噻吩吡啶盐jnld-1(盐酸盐)和jnld-15(硫酸盐)在水中的溶解性情况(见表1)。实验结果表明,丹参酮iia噻吩吡啶化合物水溶性显著提高。
表1.丹参酮iia与代表性丹参酮iia噻吩吡啶盐化合物水溶性比较
实施例26稳定性研究
稳定性是药物的重要属性之一,对此本实验利用hplc对丹参酮iia噻吩吡啶类化合物及其盐的化学稳定性进行研究。具体实验方法:将jnld-1及其盐酸盐于25℃下配制成一定浓度(0.1mg/ml)的水溶液,之后用hplc分别检测它们0、2、4、6、8、12和24小时时的254波段峰面积变化。实验结果如表2所示,jnld-1及其盐酸盐在溶液中具有优良的化学稳定性。
表2.jnld-1及其盐的稳定性
药理学实验
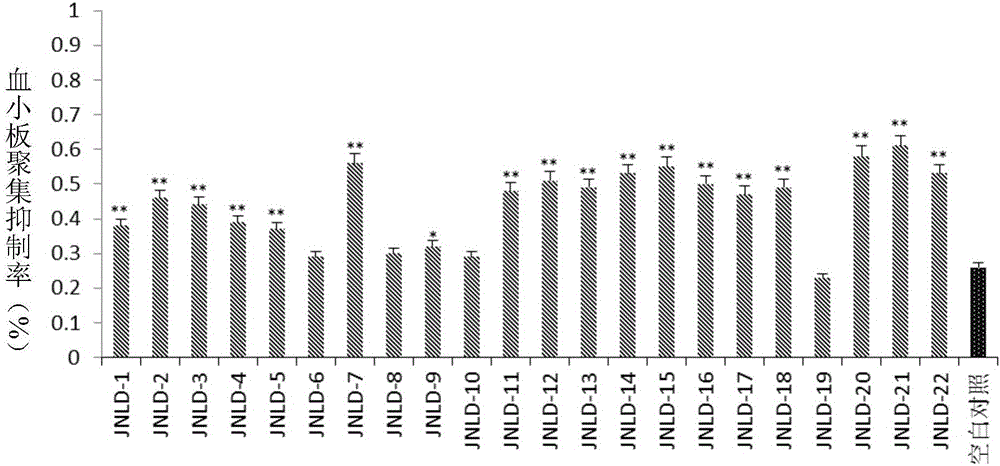
实施例27体外对adp诱导的家兔血小板聚集抑制作用研究
药品及制剂:受试化合物溶于少(定)量dmso,然后移取lμl加入100~300μl去离子水中配成10μg/ml,37℃水浴加热溶解后备用。
adp,sigma,美国
动物:雄性科研兔,2.0-2.5kg,山东大学实验动物中心
仪器:血小板聚集仪(560ca),chrono-log,美国
试验方法:雄性科研兔禁食24h,称重,5%水合氯醛麻醉,颈部动脉插管取血,109mmol/l枸橼酸钠溶液与血液以9:1的比例抗凝,收集于10ml塑料离心管中。室温条件下,900rpm离心10min,吸取上清液得prp;剰余血液以3500rpm离心10min,吸取上清液得ppp。调整prp使血小板计数为4~5×1012个/ml;取测试杯,加转子,加入lμl测试样品,300μlprp,37℃恒温5min,ppp调零后,往测试杯中加入adp,测量并记录最大聚集率。按born比浊法,用血小板聚集仪测定血小板聚集百分数,以t-检验进行统计学比较。血小板聚集抑制率按下式计算:血小板聚集抑制率(%)=[1-(给药管聚集百分率/对照管聚集百分率)]×100%。
实验结果:如附图1所示示,受试化合物在体外具有一定的抑制adp诱导的血小板聚集活性(**p<0.01vs空白对照,*p<0.05vs空白对照)。
实施例28大鼠动静脉旁路血栓形成的抑制作用研究
药品及制剂:阳性药为氯吡格雷硫酸盐。阳性药和受试化合物以0.5%cmc-na(羧甲基纤维素钠)配成混悬液供动物给药用。
实验动物:雄性sd大鼠,200-350g,共50只,由山东大学实验动物中心提供。
试验方法:旁路塑管的制作:取二根6.5cm长内径1mm的聚乙烯塑管,中间穿入一根长6cm的丝线,其中一根塑管两端用长3公分,内径约0.8mm的聚乙烯塑管相连,另一根待用,管中充满50u/ml肝素钠生理盐水待用。实验选用体重250-350g雄性sd大鼠随机分成5个剂量组。大鼠用戊巴比妥钠40mg/kg腹腔注射麻醉,麻醉大鼠仰卧固定在大鼠解剖板上,结扎食管,分离右侧颈总动脉和左侧颈外静脉。从大鼠股静脉注射50u/kg肝素钠生理盐水抗凝,然后将第一根塑料连接管两端分别插入右侧颈总动脉和左侧颈外静脉形成旁路。经灌胃给药后60min时打开动脉夹,血液从右侧颈总动脉通过旁路塑管流入左侧颈外静脉,放开血流15min后中断血流迅速取出塑管内带血栓的丝线,吸走多余血液,称丝线湿重即60min血栓形成量。将空白对照组丝线湿重和给药组丝线湿重之比求出60min时血栓形成抑制百分率并进行统计处理。
实验结果:如附图2所示,与空白对照相比,受试化合物对大鼠动静脉旁路血栓形成具有显著的抑制作用(**p<0.01vs空白对照)。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种环吡酮胺的药物组合物及其医药用图
- 一种吡噻菌胺残留量的gc-ei-ms测定方法
- 一种检测噻吩并吡啶类抗血小板药物疗效的试剂卡及其使用方法和应用
- 一种环吡酮胺的药物组合物及其医药用图
- 一种吡噻菌胺残留量的gc-ei-ms测定方法
- 一种吡噻菌胺残留量的gc-ei-ms快速测定方法
- 四氢糠醇气固相催化合成吡啶的负载型催化剂及其制备方法
- 一种合成n-取代-1,2,5,6-四氢吡啶-4-硼酸酯的方法
- (5-(2-腈基苄基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶-2-基)乙酸酯的晶型Ⅰ及其制备方 ...的制作方法
- (5-(2-腈基苄基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶-2-基)乙酸酯的晶型Ⅲ及其制备方 ...的制作方法