一种含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物的制备方法与流程
本发明涉及有机化学领域,具体涉及一种含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物的制备方法。
背景技术
含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物是制备具有α-氰基丙烯酰氧基结构有机硅化合物的重要中间体,后者可在空气中潮气作用下,通过阴离子聚合方式快速交联固化,赋予有机硅材料在温和条件下快速交联固化的能力,广泛应用于刑侦、情报收集、粘接、密封、堵漏等需要快速就地成型的领域。
文献中关于带有蒽保护的α-氰基丙烯酰氧基结构的有机硅小分子化合物的合成鲜见报道。虽然蒽保护及脱去蒽保护基团的含有α-氰基丙烯酰氧基结构的有机硅聚合物及其制备方法已有文献报道,但对有机硅聚合物进行化学改性,使有机硅聚合物的链端或者侧链接枝α-氰基丙烯酰氧基结构单元,仍然面临许多挑战。美国专利us9695286公开了一种以含有si-oh或者si-h官能团的线性聚硅氧烷为底物,通过si-oh与蒽保护的α-氰基丙烯酰氯之间的缩合反应(反应式1)或者si-h与蒽保护的α-氰基丙烯酰氧丙烯之间的硅氢加成反应(反应式2),将α-氰基丙烯酰氧基官能团接枝到聚硅氧烷的链端或者侧链的方法。
众所周知,高分子链存在的缠绕、包裹等现象会阻止聚合物分子链中的反应位点无法参与反应,导致反应转化率不高,而对于不饱和烯烃的硅氢加成反应,除了在烯烃的α位置发生加成反应外,在烯烃的b位置也存在加成反应,即存在马氏加成和反马氏加成反应之间的竞争,导致硅氢加成产物不可避免地会出现顺式和反式两种不同结构的同分异构体;另一方面,聚合物硅氢加成反应所需条件较为苛刻且产率不高,只有使用pt质量分数大于2.0wt%的karstedt类型硅氢加成催化剂时,硅氢加成反应才能进行,即便如此,反应的收率也只有50%左右。
反应式1.缩合反应制备含有蒽保护的α-氰基丙烯酰氧基结构的聚硅氧烷
反应式2.硅氢加成制备含有蒽保护的α-氰基丙烯酰氧基结构的聚硅氧烷
虽然si-oh与蒽保护的α-氰基丙烯酰氯之间的缩合反应不会产生异构体,但由于酰氯类化学品毒性较大,其半数致死量lc50的数值较小,因此蒽保护的α-氰基丙烯酰氯的制备存在一定的安全风险。
技术实现要素:
为解决现有制备含蒽保护的α-氰基丙烯酰氧基结构的有机硅聚合物存在反应条件苛刻、副产物多且目标产物收率较低的问题,本发明提出了一种含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物的制备方法,具有能耗低、环境污染小、安全环保、产率高、产物容易分离、工艺及设备简单且生产成本低等特点。
所述的含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物,其化学结构式如(i)所示,
式中,r1,r2,r3分别独立选自r或or基团,r选自甲基、乙基、正丙基、异丙基、正丁基、异丁基中一种。
本发明是通过下述技术方案得以实现的:一种含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物的制备方法采用两步法制备得到:
(1)将γ-氯丙基硅烷与蒽保护的α-氰基丙烯酸钾盐加入到非质子极性溶剂中,搅拌混合,然后向反应体系中加入卤代反应催化剂和酯化反应催化剂;加料完毕后,将物料升温并在该温度下进行反应;然后降至室温后,将反应产物过滤,收集滤液,得到含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物溶液;
所述的γ-氯丙基硅烷选自γ-氯丙基三烷基硅烷、γ-氯丙基烷基二烷氧基硅烷、γ-氯丙基二烷基烷氧基硅烷、γ-氯丙基三烷氧基硅烷中的一种,其结构如式(ii)所示,
式中,r1,r2,r3分别独立选自烷基r或烷氧基or,r选自甲基、乙基、正丙基、异丙基、正丁基、异丁基中一种;
作为优选,所述的γ-氯丙基硅烷选自γ-氯丙基烷氧基硅烷;更优选,所述的γ-氯丙基硅烷选自γ-氯丙基三甲氧基硅烷,γ-氯丙基三乙氧基硅烷,γ-氯丙基甲基二甲氧基硅烷或γ-氯丙基甲基二乙氧基硅烷中的一种。
所述的γ-氯丙基硅烷与蒽保护的α-氰基丙烯酸钾盐的摩尔比为0.5~5∶1,作为优选,γ-氯丙基硅烷与蒽保护的α-氰基丙烯酸钾盐的摩尔比为0.7~2∶1,更优选,γ-氯丙基硅烷与蒽保护的α-氰基丙烯酸钾盐的摩尔比为0.95~1.15∶1。
所述的非质子极性溶剂选自二甲亚砜(dmso),乙腈,n,n-二甲基甲酰胺(dmf),n,n-二甲基乙酰胺(dmac),丙酮,六甲基磷酰胺(hmp)中的一种或几种;作为优选,所述的非质子极性溶剂选自dmf、dmac中的一种或两种;更优选,所述的非质子极性溶剂选自dmf。
所述的非质子极性溶剂的用量为蒽保护的α-氰基丙烯酸钾盐质量的3~15倍。作为优选,所述的非质子极性溶剂的用量为蒽保护的α-氰基丙烯酸钾盐质量的5~10倍;更优选,所述的非质子极性溶剂的用量为蒽保护的α-氰基丙烯酸钾盐质量的6~9倍。
所述的卤代反应催化剂选自lif、licl、libr、lii、naf、nacl、nabr、nai、kf、kcl、kbr、ki中的一种;作为优选,所述的卤代反应催化剂选自kbr或ki中的一种。
所述卤代反应催化剂的用量为蒽保护的α-氰基丙烯酸钾盐质量的5%~25%。作为优选,所述卤代反应催化剂的用量为蒽保护的α-氰基丙烯酸钾盐质量的7.5%~15%。
所述的酯化反应催化剂选自2-氨基吡啶、3-氨基吡啶、4-氨基吡啶、2-二甲氨基吡啶、3-二甲氨基吡啶、4-二甲氨基吡啶、2-(2-氨乙基)吡啶中的一种;作为优选,所述的酯化反应催化剂选自2-二甲氨基吡啶,3-二甲氨基吡啶,4-二甲氨基吡啶中的一种;更优选,所述的酯化反应催化剂选自4-二甲氨基吡啶。
所述的酯化反应催化剂的用量为蒽保护的α-氰基丙烯酸钾盐质量的0.1%~10%;作为优选,所述的酯化反应催化剂的用量是蒽保护的α-氰基丙烯酸钾盐质量的0.5%~5%。
所述反应温度为100~140℃,反应时间为0.5~5h,作为优选,温度为110~130℃,反应时间为1~4h。
(2)将含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物溶液缓慢滴加到氯化钠水溶液中,滴加完毕后静置静置2~4h,待产物沉降完毕后,去除水层,回收有机层;将有机层溶解在卤代烷烃中,使用去离子水反复洗涤直至水相澄清且无色透明,然后回收有机层;采用无水硫酸钠脱除有机层夹带的水分后,过滤,将收集的滤液采用减压蒸馏等方式脱除溶剂后,得到含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物。
所述的氯化钠水溶液中氯化钠的质量百分含量为3%~25%;作为优选,所述的氯化钠水溶液中氯化钠的质量百分含量为5%~20%;更优选,所述的氯化钠水溶液中氯化钠质量百分含量为5%~15%。
所述的氯化钠水溶液的用量为蒽保护的α-氰基丙烯酸钾盐质量的20~60倍。作为优选,所述的氯化钠水溶液的用量为蒽保护的α-氰基丙烯酸钾盐质量的25~50倍;更优选为30~40倍。
所述的卤代烷烃选自一取代的卤代烷烃、二取代的卤代烷烃、三取代的卤代烷烃、四取代的卤代烷烃中的一种,作为优选,所述的卤代烷烃选自二取代的卤代烷烃;其中,所述的烷烃选自甲烷、乙烷、丙烷、丁烷、戊烷、己烷中的一种,作为优选,所述的烷烃为甲烷或乙烷;更优选,所述的卤代烷烃为二氯甲烷。
所述的卤代烷烃的用量为蒽保护的α-氰基丙烯酸钾盐质量的3~50倍,作为优选,所述的卤代烷烃的用量为蒽保护的α-氰基丙烯酸钾盐质量的5~40倍;更优选为7~30倍。
本发明的制备方法在是室温下进行的,20℃±5℃。
本发明以γ-氯丙基硅烷和蒽保护的α-氰基丙烯酸钾盐为原料,利用缩合反应制备得到含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物,反应式如下所示:
由本发明制备的含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物出发,通过自身的水解、缩合等反应或与其他有机硅单体一起进行共水解、缩合等反应,可以制备得到带有蒽保护的α-氰基丙烯酰氧基结构的有机硅聚合物,后者脱除蒽保护基团后,可以得到快速交联固化的有机硅聚合物;或者,先将本发明制备的含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物脱去蒽保护基团后,再通过自身的水解、缩合等反应或与其他有机硅单体一起进行共水解、缩合等反应,也可以制备得到快速交联固化的有机硅聚合物。
与现有技术相比,本发明的有益效果是:反应条件温和,产物组成单一,含有蒽保护的α-氰基丙烯酰氧基结构的有机硅化合物合成收率高,易于分离纯化,易于实现产业化。
附图说明
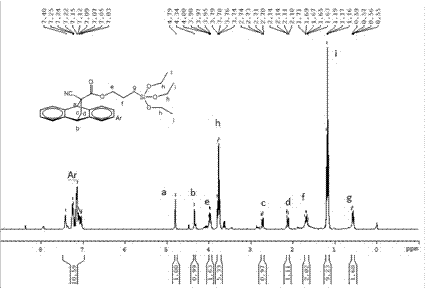
图1为蒽保护的α-氰基丙烯酰氧丙基三乙氧基硅烷的1h-nmr谱图;
图2为蒽保护的α-氰基丙烯酰氧丙基甲基二乙氧基硅烷的1h-nmr谱图。
具体实施方式
下面结合实施例对本发明作进一步详细说明,但实施例不是对本发明保护范围的限制。实施例中所用原料均可市购或采用常规方法制备。
实施例1
在100ml装有冷凝管、温度计和电磁搅拌的三口烧瓶中加入20mldmf溶剂,室温搅拌下加入3.13g(0.01mol)蒽保护的α-氰基丙烯酸钾盐,2.48g(0.0103mol)γ-氯丙基三乙氧基硅烷,0.311g卤代反应催化剂碘化钾,0.0319g酯化反应催化剂4-二甲氨基吡啶,然后升温至120℃,在此温度下反应2h后降至室温,反应粗产物过滤除去副产物kcl后,将收集的滤液以每秒1-2滴的速度缓慢滴加到搅拌状态下的100ml质量分数为12%的氯化钠水溶液中,滴加完毕后静置2h,待产物沉降后,分液除去上层水层,向收集的有机层中加入40ml的二氯甲烷溶解产物,再在二氯甲烷溶液中加入80ml去离子水,洗去产物中残留的dmf溶剂,然后再次分液去除水层保留有机层,重复7次,直到反应水层澄清透明,去除水层,向收集的二氯甲烷溶液有机层中加入8.54g无水硫酸钠干燥滤液,静置5h后,过滤,将收集的滤液进行减压蒸馏,得到蒽保护的α-氰基丙烯酰氧丙基三乙氧基硅烷4.52g,产率为92.3%。
实施例2
在250ml装有冷凝管、温度计和电磁搅拌的三口烧瓶中加入50mldmac(n,n-二甲基乙酰胺)溶剂,室温搅拌下加入3.13g(0.01mol)蒽保护的α-氰基丙烯酸钾盐,11.91g(0.0495mol)γ-氯丙基三乙氧基硅烷,0.783g卤代反应催化剂溴化钾,0.1565g酯化反应催化剂2-二甲氨基吡啶,然后升温至120℃,在此温度下反应5h后降至室温,反应粗产物过滤除去副产物kcl后,将收集的滤液以每秒1-2滴的速度缓慢滴加到搅拌状态下的180ml质量分数为13%的氯化钠水溶液中,滴加完毕后静置2h,待产物沉降后,分液除去上层水层,向收集的有机层中加入110ml的二氯甲烷溶解产物,再在二氯甲烷溶液中加入180ml去离子水,洗去产物中残留的dmac溶剂,然后再次分液去除水层保留有机层,重复8次,直到反应水层澄清透明,去除水层,向收集的二氯甲烷溶液有机层中加入20g无水硫酸钠干燥滤液,静置5h后,过滤,将收集的滤液进行减压蒸馏,得到蒽保护的α-氰基丙烯酰氧丙基三乙氧基硅烷4.29g,产率为89.7%。
实施例3
在100ml装有冷凝管、温度计和电磁搅拌的三口烧瓶中加入20mldmso(二甲亚砜)溶剂,室温搅拌下加入3.13g(0.01mol)蒽保护的α-氰基丙烯酸钾盐,2.98g(0.015mol)γ-氯丙基三甲氧基硅烷,0.157g卤代反应催化剂碘化钠,0.017g酯化反应催化剂4-氨基吡啶,然后升温至100℃,在此温度下反应5h后降至室温,反应粗产物过滤除去副产物kcl后,将收集的滤液以每秒1-2滴的速度缓慢滴加到搅拌状态下的70ml质量分数为9%的氯化钠水溶液中,滴加完毕后静置6h,待产物沉降后,分液除去上层水层,向收集的有机层中加入20ml的三氯甲烷溶解产物,再在三氯甲烷溶液中加入90ml去离子水,洗去产物中残留的dmso溶剂,然后再次分液去除水层保留有机层,重复7次,直到反应水层澄清透明,去除水层,向收集的三氯甲烷溶液有机层中加入18.70g无水硫酸钠干燥滤液,静置5h后,过滤,将收集的滤液进行减压蒸馏,得到蒽保护的α-氰基丙烯酰氧丙基三甲氧基硅烷3.82g,产率为87.4%。
实施例4
在50ml装有冷凝管、温度计和电磁搅拌的三口烧瓶中加入10mldmf溶剂,室温搅拌下加入1.57g(0.005mol)蒽保护的α-氰基丙烯酸钾盐,1.22g(0.0067mol)γ-氯丙基甲基二甲氧基硅烷,0.156g卤代反应催化剂碘化钾,0.0056g酯化反应催化剂2-(2-氨乙基)吡啶,然后升温至120℃,在此温度下反应2h后降至室温,反应粗产物过滤除去副产物kcl后,将收集的滤液以每秒1-2滴的速度缓慢滴加到搅拌状态下的80ml质量分数为12%的氯化钠水溶液中,滴加完毕后静置4h,待产物沉降后,分液除去上层水层,向收集的有机层中加入60ml的1,2-二氯乙烷溶解产物,再在1,2-二氯乙烷溶液中加入160ml去离子水,洗去产物中残留的dmf溶剂,然后再次分液去除水层保留有机层,重复8次,直到反应水层澄清透明,去除水层,向收集的1,2-二氯乙烷溶液有机层中加入5.57g无水硫酸钠干燥滤液,静置5h后,过滤,将收集的滤液进行减压蒸馏,得到蒽保护的α-氰基丙烯酰氧丙基甲基二甲氧氧基硅烷1.92g,产率为91.11%。
实施例5
在250ml装有冷凝管、温度计和电磁搅拌的三口烧瓶中加入40ml六甲基磷酰胺溶剂,室温搅拌下加入13.13g(0.042mol)蒽保护的α-氰基丙烯酸钾盐,12.66g(0.084mol)γ-氯丙基三甲基硅烷,1.315g卤代反应催化剂碘化钾,0.1314g酯化反应催化剂4-氨基吡啶,然后升温至130℃,在此温度下反应4.5h后降至室温,反应粗产物过滤除去副产物kcl后,将收集的滤液以每秒1-2滴的速度缓慢滴加到搅拌状态下的400ml质量分数为7%的氯化钠水溶液中,滴加完毕后静置2h,待产物沉降后,分液除去上层水层,向收集的有机层中加入40ml的二氯甲烷溶解产物,再在二氯甲烷溶液中加入160ml去离子水,洗去产物中残留的六甲基磷酰胺溶剂,然后再次分液去除水层保留有机层,重复9次,直到反应水层澄清透明,去除水层,向收集的二氯甲烷溶液有机层中加入31.76g无水硫酸钠干燥滤液,静置5h后,过滤,将收集的滤液进行减压蒸馏,得到蒽保护的α-氰基丙烯酰氧丙基三甲基硅烷14.91g,产率为91.13%。
实施例6
在50ml装有冷凝管、温度计和电磁搅拌的三口烧瓶中加入10ml六甲基磷酰胺溶剂,室温搅拌下加入1.57g(0.005mol)蒽保护的α-氰基丙烯酸钾盐,0.542g(0.003mol)γ-氯丙基二甲基乙氧基硅烷,0.156g卤代反应催化剂溴化锂,0.0158g酯化反应催化剂3-氨基吡啶,然后升温至110℃,在此温度下反应4h后降至室温,反应粗产物过滤除去副产物kcl后,将收集的滤液以每秒1-2滴的速度缓慢滴加到搅拌状态下的60ml质量分数为25%的氯化钠水溶液中,滴加完毕后静置2h,待产物沉降后,分液除去上层水层,向收集的有机层中加入35ml的四氯化碳溶解产物,再在四氯化碳溶液中加入70ml去离子水,洗去产物中残留的六甲基磷酰胺溶剂,然后再次分液去除水层保留有机层,重复7次,直到反应水层澄清透明,去除水层,向收集的四氯化碳溶液有机层中加入6.69g无水硫酸钠干燥滤液,静置5h后,过滤,将收集的滤液进行减压蒸馏,得到蒽保护的α-氰基丙烯酰氧丙基二甲基乙氧基硅烷1.79g,产率为85.4%。
实施例7
在100ml装有冷凝管、温度计和电磁搅拌的三口烧瓶中加入20mldmf溶剂,室温搅拌下加入2.82g(0.009mol)蒽保护的α-氰基丙烯酸钾盐,2.17g(0.009mol)γ-氯丙基三乙氧基硅烷,0.173g卤代反应催化剂碘化钾,0.0173g酯化反应催化剂4-二甲氨基吡啶,然后升温至120℃,在此温度下反应1h后降至室温,反应粗产物过滤除去副产物kcl后,将收集的滤液以每秒1-2滴的速度缓慢滴加到搅拌状态下的100ml质量分数为6%的氯化钠水溶液中,滴加完毕后静置2h,待产物沉降后,分液除去上层水层,向收集的有机层中加入45ml的二氯甲烷溶解产物,再在二氯甲烷溶液中加入90ml去离子水,洗去产物中残留的dmf溶剂,然后再次分液去除水层保留有机层,重复9次,直到反应水层澄清透明,去除水层,向收集的二氯甲烷溶液有机层中加入7.53g无水硫酸钠干燥滤液,静置5h后,过滤,将收集的滤液进行减压蒸馏,得到蒽保护的α-氰基丙烯酰氧丙基三乙氧基硅烷3.53g,产率为81.8%。
实施例8
在100ml装有冷凝管、温度计和电磁搅拌的三口烧瓶中加入20mldmf溶剂,室温搅拌下加入3.13g(0.01mol)蒽保护的α-氰基丙烯酸钾盐,2.11g(0.01mol)γ-氯丙基甲基二乙氧基硅烷,0.316g卤代反应催化剂碘化钾,0.0310g酯化反应催化剂4-二甲氨基吡啶,然后升温至120℃,在此温度下反应2h后降至室温,反应粗产物过滤除去副产物kcl后,将收集的滤液以每秒1-2滴的速度缓慢滴加到搅拌状态下的100ml质量分数为7%的氯化钠水溶液中,滴加完毕后静置2h,待产物沉降后,分液除去上层水层,向收集的有机层中加入45ml的二氯甲烷溶解产物,再在二氯甲烷溶液中加入100ml去离子水,洗去产物中残留的dmf溶剂,然后再次分液去除水层保留有机层,重复8次,直到反应水层澄清透明,去除水层,向收集的二氯甲烷溶液有机层中加入9.08g无水硫酸钠干燥滤液,静置5h后,过滤,将收集的滤液进行减压蒸馏,得到蒽保护的α-氰基丙烯酰氧丙基甲基二乙氧基硅烷4.08g,产率为90.7%。
实施例1,2和7制备的蒽保护的α-氰基丙烯酰氧丙基三乙氧基硅烷的1h-nmr谱图如图1所示。实施例8制备的蒽保护的α-氰基丙烯酰氧丙基甲基二乙氧基硅烷的1h-nmr谱图如图2所示。
- 还没有人留言评论。精彩留言会获得点赞!