一种生物碱磷酸盐阻燃剂及其制备方法与流程
本发明属于高分子材料技术领域,具体涉及一种生物碱磷酸盐阻燃剂及其制备方法。
背景技术:
随着高分子材料的发展,聚烯烃材料已经广泛应用于日常生活和工业领域。但由于其本身结构原因,使其具有易燃的特性,这也给人们带来了潜在的安全隐患。为了降低聚烯烃类材料易燃带来的安全隐患,必须对其进行阻燃改性。
传统的阻燃剂有卤系阻燃剂和无卤阻燃剂。卤系阻燃剂阻燃效果好,添加量少,但存在发烟量大,释放有毒物质较多等缺点。而无卤阻燃剂能很好的避免卤素所产生的有毒物质,因此越来越多地受到人们的关注。
其中膨胀型阻燃剂因其在燃烧过程中低烟,低毒,无腐蚀性气体产生等特点,符合阻燃剂的绿色化的要求,已经成了重点研究的方向之一。膨胀型阻燃剂一般含有酸源、碳源、气源三种组分,它们在燃烧过程中相互作用产生多孔炭层,起到隔热隔氧的功效,因而发挥阻燃作用。
此外,随着技术的发展,人们不但要求阻燃剂本身环保,对其来源及可持续性也有了新的考虑。采用生物原料制备阻燃剂因而受到大量关注。但是大部分生物原料本身存在很多缺陷,无法作为良好的阻燃剂使用,必须对其结构进行设计和改善,赋予其作为阻燃剂使用的性能。很多原料已经被尝试用于阻燃材料中,如壳聚糖、纤维素、淀粉、环糊精等,这类材料由于其不含磷元素,可以用来做成炭剂,或经改性后作为阻燃剂使用。xiaofengwang(ind.eng.chem.res.2013(52):3287-3294)等人通过主客体作用以β-环糊精与聚丙二醇为原料制备了聚轮烷,并将其与聚磷酸铵(app)、三聚氰胺(ma)复配阻燃聚乳酸(pla)。结果表明,材料的氧指数(loi)值明显提高,总热释放明显降低,说明材料获得了良好的阻燃性能。weizhaohu(j.therm.anal.calorim.2014(117):27-38)等人用乙基纤维素改性app阻燃聚丁二酸二丁酯,结果表明改性app和成炭剂的加入可以明显提高材料的loi,降低材料的峰值热释放速率。wangjingjing(ind.eng.chem.res.2014(53):1422-1430)等将淀粉与含磷阻燃剂复配添加到pla中,结果表明,固定含磷阻燃剂的添加量,3%的淀粉可以使pla复合材料的loi提高到33.0vol%。galinalaufer(acsappl.mater.interfaces,2012(4):670-676)等人通过层层自组装的方法将壳聚糖和粘土涂覆与聚氨酯表面,结果表明该涂层能大大降低聚氨酯的热释放峰值。这些不含磷的生物基材料往往只能与含磷阻燃剂复配使用。除此之外一些含磷的生物基材料也被应用于阻燃例如脱氧核糖核酸(dna)、核苷酸等。boscofrancesca(surf.coat.technol.2015(272):86-95)等人将dna通过层层自组装技术涂覆于棉织物表面,结果表明,dna涂层赋予了棉织物优异的阻燃性能。wang,zhijing(j.anal.at.pyrolysis,2016(121):394-402)等人制备了三聚氰胺甲醛树脂包覆的核苷酸(mfa),将其与膨胀阻燃剂协同阻燃聚丙烯,结果表明添加17wt%ifr和1wt%mfa可以使聚丙烯复合材料通过ul-94v-0等级测试。这些含磷的生物基阻燃剂虽然显示了良好的阻燃效果,但由于其热稳定性较差、阻燃效率低等原因限制了其应用。
据报道,酸碱催化对成炭往往有促进作用。自然界有许多生物碱,它们热稳定性好,能与磷酸形成盐,而且还具有一定的催化作用,有望形成具有催化阻燃作用的新型阻燃剂,相关研究还未见公开报道。本专利采用生物碱和磷酸反应形成生物碱磷酸盐阻燃剂,并对其制备方法进行优化。
技术实现要素:
本发明的一个目的是针对现有生物基膨胀型阻燃剂的不足,提出一种生物碱磷酸盐阻燃剂。该生物基阻燃剂,在保证阻燃性能的前提下,降低阻燃剂的添加量,提高阻燃效率。
本发明阻燃剂采用生物碱单体x、磷酸类单体y通过反应形成生物碱磷酸盐,然后采用环氧树脂或聚氨酯对其进行表面包覆改性处理得到,提高其耐水性及其与聚合物的相容性。
所述生物碱单体x为胞嘧啶(c)、尿嘧啶(u)、腺嘌呤(a)、鸟嘌呤(g)、胸腺嘧啶(t)中的一种或多种,其结构式分别如下:
所述磷酸类单体y为:焦磷酸,过磷酸、多聚磷酸,苯膦酸,植酸。
本发明的另一个目的是提供上述生物碱磷酸盐阻燃剂的制备方法,包括下述工艺步骤:
步骤(1)、阻燃剂xy制备:
将生物碱单体x分散在溶剂z中,加热至40-120℃,得到生物碱单体x溶液;磷酸类单体y溶解在溶剂d中,得到磷酸类单体y溶液;然后在惰性气体保护下将磷酸类单体y溶液滴加到生物碱单体x溶液中,搅拌反应2-48h,抽滤,洗涤,干燥,得到阻燃剂xy。
所述的生物碱单体x与磷酸类单体y的摩尔比为1-4:1。
步骤(2)、阻燃剂xy的表面处理方法:
第一种:将步骤(1)所得的阻燃剂xy分散在溶剂z中,混匀得到阻燃剂xy溶液;化合物e溶解在溶剂d中,混匀得到化合物e溶液;然后将固化剂f和化合物e溶液添加到上述阻燃剂xy溶液中,在80-170℃下反应6-48h,抽滤,洗涤,干燥得到表面处理后的阻燃剂m1-xy。其中化合物e和固化剂f的摩尔比为1-10:1,阻燃xy与包覆材料的质量比为2-10:1。
第二种:将步骤(1)所得的阻燃剂xy分散在溶剂z中,得阻燃剂xy溶液;化合物h溶解在溶剂d中,得到化合物h溶液,搅拌;然后将固化剂i、催化剂j和化合物h溶液添加到阻燃剂xy溶液,在80-170℃下反应6-48h,抽滤,洗涤,干燥,得到表面处理后的阻燃剂m2-xy。其中化合物h和固化剂i的摩尔比为1-10:1。催化剂j的用量为0.001-0.01,阻燃xy与包覆材料的质量比为2-10:1。
所述的溶剂z为甲醇、乙醇、四氢呋喃、二氧六环、石油醚、吡啶、丙酮、丁酮、氯仿、二氯甲烷、二氯乙烷、氯化亚砜、四氯化碳、二硫化碳、dmf、dmso、苯、甲苯、二甲苯、硝基苯、氯苯、乙腈、环己烷或正己烷中的一种或几种的混合溶剂;
所述的溶剂d为甲醇、乙醇、四氢呋喃、二氧六环、石油醚、吡啶、丙酮、丁酮、氯仿、二氯甲烷、二氯乙烷、氯化亚砜、四氯化碳、二硫化碳、dmf、dmso、苯、甲苯、二甲苯、硝基苯、氯苯、乙腈、环己烷或正己烷中的一种或几种的混合溶剂;
所述的化合物e为环氧cyd-128、环氧cyd-134、环氧cyd-118、环氧cyd-546、异氰尿酸三缩水甘油酯中的一种。
所述的化合物f为乙二胺、二乙烯三胺、三乙烯四胺、二丙烯三胺、二甲氨基丙胺、二乙氨基丙胺、三甲基六亚甲基二胺、4,4-二氨基二苯甲烷、间苯二胺、邻苯二甲酸酐、均苯四甲酸酐中的一种。
所述的化合物h为甲苯二异氰酸酯(tdi)、异佛尔酮二异氰酸酯(ipdi)、二苯基甲烷二异氰酸酯(mdi)、二环己基甲烷二异氰酸酯(hmdi)、六亚甲基二异氰酸酯(hdi)、赖氨酸二异氰酸酯(ldi)中的一种。
所述固化剂i为乙二胺、二乙烯三胺、三乙烯四胺、二甲氨基丙胺、二乙氨基丙胺、三甲基六亚甲基二胺、间苯二胺、季戊四醇、甘油、三羟甲基乙烷、木糖醇、山梨醇中的一种。
所述催化剂j为二月桂酸二丁基锡、辛酸亚锡中的一种。
本发明的有益效果为:通过生物基化合物碱基为原料,制备了一系列含磷碱基盐,该含磷碱基盐能够作为阻燃添加剂使用,而且阻燃效率较高,为提高生物基阻燃添加剂的阻燃效率提供一种可行的实施方案。
附图说明
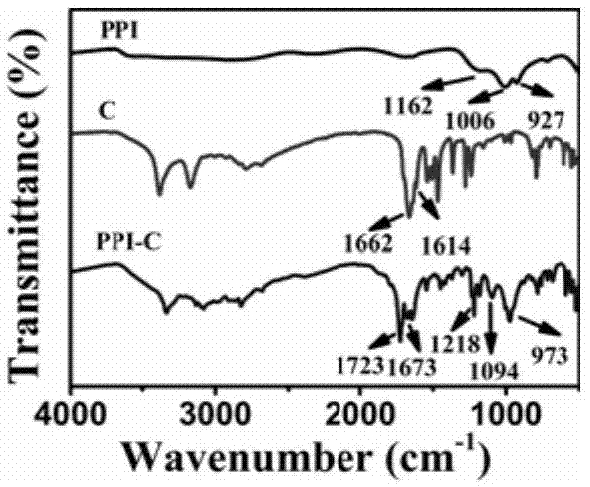
图1为实施例1-1焦磷酸(ppi),胞嘧啶(c)和产物1(ppi-c)的红外光谱图;
图2为实施例1-1胞嘧啶(c)和产物1(ppi-c)在氮气下的热失重曲线;
图3为实施例1-3焦磷酸(ppi),尿嘧啶(u)和产物3(ppi-u)的红外谱图;
图4为实施例1-3尿嘧啶(u)和产物3(ppi-u)在氮气下的热失重曲线;
图5为实施例1-5焦磷酸(ppi),腺嘌呤(a)和产物5(ppi-a)的红外谱图;
图6为实施例1-5腺嘌呤(a)和产物5(ppi-a)在氮气下的热失重曲线;
图7为实施例1-7焦磷酸(ppi),鸟嘌呤(g)和产物7(ppi-g)的红外谱图;
图8为实施例1-7鸟嘌呤(g)和产物7(ppi-g)在氮气下的热失重曲线。
具体实施方式
本发明提到的上述特征,或具体实例提到的特征可以任意组合。下面结合实例,对本发明做进一步详细阐述,这些具体实施例子仅用于说明本发明而不用于限制本发明的范围。
本发明采用红外光谱仪和热重分析仪对实施例中所得产品进行表征。
下述实施例制备得到的产品均落入本发明的要求。
实例1-1
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的100ml三口圆底烧瓶中,加入乙醇(30ml,已除水)、胞嘧啶(2.22g,0.02mol)。氮气条件下搅拌并升温至60℃。称焦磷酸(1.78g,0.01mol)加入烧杯中,加乙醇(20ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到3.84g白色固体产物1(产率为:
y%=96.0%),产物结构见式1。图1为实施例1-1产物的红外光谱图。图2为实施例1-1产物热重曲线。
实例1-2
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入甲醇(200ml,已除水)、胞嘧啶(22.22g,0.20mol)。氮气条件下搅拌并升温至60℃。称取焦磷酸(8.90g,0.05mol)加入烧杯中,加甲醇(50ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应4h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到29.56g白色固体产物2(产率为:y%=95.0%),产物结构见式2。
实例1-3
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的250ml三口圆底烧瓶中,加入乙腈(100ml,已除水)、尿嘧啶(11.2g,0.1mol)。氮气条件下搅拌并升温至90℃。称取焦磷酸(8.90g,0.05mol)加入烧杯中,加乙腈(20ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应8h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到18.24g白色固体产物3(产率为:y%=90.7%),产物结构见式3。图3为实施例1-3产物的红外光谱图;图4为实施例1-3产物热重曲线。
实例1-4
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的100ml三口圆底烧瓶中,加入四氢呋喃(40ml,已除水)、尿嘧啶(4.48g,0.04mol)。氮气条件下搅拌并升温至80℃。称取焦磷酸(1.78g,0.01mol)加入烧杯中,加四氢呋喃(20ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应12h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到5.14g白色固体产物4(产率为:y%=82.1%)产物结构见式4。
实例1-5
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的1l三口圆底烧瓶中,加入二氧六环(300ml,已除水)、腺嘌呤(27.02g,0.20mol)。氮气条件下搅拌并升温至95℃。称取焦磷酸(17.80g,0.10mol)加入烧杯中,加二氧六环(100ml,已除水)搅拌至完全溶解,然后通过滴液漏斗分两次缓慢滴加到三口烧瓶中搅拌反应16h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到38.64g白色固体产物5(产率为:y%=86.2%),产物结构见式5。图5为实施例1-5产物的红外光谱图;图6为实施例1-5产物热重曲线。
实例1-6
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入dmf(200ml,已除水)、腺嘌呤(10.81g,0.08mol)。氮气条件下搅拌并升温至120℃。称取焦磷酸(3.56g,0.02mol)加入烧杯中,加dmf(100ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应9h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到12.54g白色固体产物6(产率为:y%=87.3%),产物结构见式6。
实例1-7
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的100ml三口圆底烧瓶中,加入二氯甲烷(30ml,)、鸟嘌呤(3.02g,0.02mol)。氮气条件下搅拌并升温至40℃。称取焦磷酸(1.78g,0.01mol)加入烧杯中,加二氯甲烷(20ml,)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应5h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到3.97g白色固体产物7(产率为:y%=82.7%),产物结构见式7。
实例1-8
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入氯仿(250ml)、鸟嘌呤(30.22g,0.20mol)。氮气条件下搅拌并升温至60℃。称取焦磷酸(8.90g,0.05mol)加入烧杯中,加氯仿(80ml)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到34.21g白色固体产物8(产率为:y%=87.4%),产物结构见式8。
实例1-9
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的250ml三口圆底烧瓶中,加入乙醇(100ml,已除水)、胸腺嘧啶(10.09g,0.08mol)。氮气条件下搅拌并升温至75℃。称取焦磷酸(7.12g,0.04mol)加入烧杯中,加乙醇(80ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应14h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到15.37g白色固体产物9(产率为:y%=89.3%),产物结构见式9。
实例1-10
在装有球形冷凝管、加热机械搅拌,温度计及滴液漏斗的2l三口圆底烧瓶中,加入甲醇(700ml,已除水)、胸腺嘧啶(50.44g,0.40mol)。氮气条件下搅拌并升温至60℃。称取焦磷酸(17.80g,0.10mol)加入烧杯中,加乙醇(300ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到61.75g白色固体产物10(产率为:y%=90.5%),产物结构见式10。
实例1-11
在装有球形冷凝管、加热机械搅拌,温度计及滴液漏斗的5l三口圆底烧瓶中,加入乙醇(1.5l,已除水)、胞嘧啶(111.10g,1mol)。氮气条件下搅拌并升温至60℃。称取焦磷酸(177.98g,1mol)加入烧杯中,加乙醇(1.5l,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到263.83g白色固体产物11(产率为:y%=91.3%),产物结构见式11。
实例1-12
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的250ml三口圆底烧瓶中,加入乙醇(100ml,已除水)、胞嘧啶(9.99g,0.09mol)、。氮气条件下搅拌并升温至60℃。称取焦磷酸(5.34g,0.03mol)加入烧杯中,加乙醇(50ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应8h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到14.31g白色固体产物12(产率为:y%=93.3%),产物结构见式12。
实例1-13
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入乙醇(200ml,已除水)、胞嘧啶(11.11g,0.10mol)、腺嘌呤(13.51g,0.10mol)。氮气条件下搅拌并升温至60℃。称取焦磷酸(17.80g,0.10mol)加入烧杯中,加乙醇(120ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到37.35g白色固体产物13(产率为:y%=88.0%),产物结构见式13。
实例1-14
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的250ml三口圆底烧瓶中,加入乙醇(100ml,已除水)、胞嘧啶(3.33g,0.03mol)、腺嘌呤(4.05g,0.03mol)、尿嘧啶(3.36g,0.03mol)。氮气条件下搅拌并升温至60℃。称取焦磷酸(5.34g,0.03mol)加入烧杯中,加乙醇(50ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到13.86g白色固体产物14(产率为:y%=86.2%),产物结构见式14。
实例1-15
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的250ml三口圆底烧瓶中,加入甲醇(100ml,已除水)、胞嘧啶(2.22g,0.02mol)、腺嘌呤(2.70g,0.02mol)、尿嘧啶(2.24g,0.02mol)、胸腺嘧啶(2.52,0.02mol)。氮气条件下搅拌并升温至60℃。称取焦磷酸(3.56g,0.02mol)加入烧杯中,加乙醇(50ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到11.67g白色固体产物15(产率为:y%=88.1%),产物结构见式15。
实例1-16
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入甲醇(200ml,已除水)、胞嘧啶(20.00g,0.18mol)。氮气条件下搅拌并升温至60℃。称取三聚磷酸(6.84g,0.06mol)加入烧杯中,加甲醇(50ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到22.49g白色固体产物16(产率为:y%=83.8%),产物结构见式16。
实例1-17
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入甲醇(200ml,已除水)、胞嘧啶(20.00g,0.18mol)。氮气条件下搅拌并升温至60℃。称取四聚磷酸(10.14g,0.03mol)加入烧杯中,加甲醇(50ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到25.68g白色固体产物17(产率为:y%=85.2%),产物结构见式17。
实例1-18
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入甲醇(200ml,已除水)、胞嘧啶(22.22g,0.20mol)。氮气条件下搅拌并升温至60℃。称取苯膦酸(15.81g,0.10mol)加入烧杯中,加甲醇(50ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到32.18g白色固体产物18(产率为:y%=84.6%),产物结构见式18。
实例1-19
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入甲醇(200ml,已除水)、胞嘧啶(26.67g,0.24mol)。氮气条件下搅拌并升温至60℃。称取植酸(13.20g,0.02mol)加入烧杯中,加甲醇(50ml,已除水)搅拌至完全溶解,然后通过滴液漏斗缓慢滴加到三口烧瓶中搅拌反应6h。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得到36.43g白色固体产物19(产率为:y%=91.4%),产物结构见式19。
实例2-1
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入dmf(200ml,已除水)、产物1(28.91g,0.10mol),搅拌并升温至120℃。称取4,4`-二苯基甲烷二异氰酸酯(7.51g,0.03mol)、三乙烯四胺(1.46g,0.01mol)加入烧杯中,加dmf(20ml,已除水)搅拌至完全溶解,加入到上述溶液。滴加4滴催化剂二月桂酸二丁基锡。通氮气2分钟,然后抽真空,再通氮气,反复2次,氮气后将体系密闭。在120℃下反应12小时。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得36.06g包覆后的淡黄色固体产物,其结构式见式20(产率为:95.2%),见式20。
实例21
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的1l三口圆底烧瓶中,加入dmf(400ml,已除水)、产物1(57.82g,0.20mol),搅拌并升温至110℃。称取甲苯-2,4-二异氰酸酯(10.45g,0.06mol)、间苯二胺(3.24g,0.03mol)加入烧杯中,加dmf(50ml,已除水)搅拌至完全溶解,加入到上述溶液。滴加10滴催化剂辛酸亚锡。通氮气2分钟,然后抽真空,再通氮气,反复2次,氮气后将体系密闭。在110℃下反应8小时。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得66.93g包覆后的淡黄色固体产物,其结构式见式21(产率为:93.6%)。
实例22
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入dmf(200ml,已除水)、产物1(43.37g,0.15mol),搅拌并升温至120℃。称取异氰尿酸三缩水甘油酯(8.92g,0.03mol)、三乙烯四胺(2.92g,0.02mol)加入烧杯中,加dmf(50ml,已除水)搅拌至完全溶解,加入到上述溶液。在120℃下反应12小时。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得53.06g包覆后的淡黄色固体产物,其结构式见式22(产率为:96.1%)。
实例23
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的1l三口圆底烧瓶中,加入dmf(500ml,已除水)、产物1(57.82g,0.20mol),搅拌并升温至120℃。称取异氰尿酸三缩水甘油酯(17.84g,0.06mol)、乙二胺(2.40g,0.04mol)加入烧杯中,加dmf(50ml,已除水)搅拌至完全溶解,加入到上述溶液。在120℃下反应12小时。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得75.95g包覆后的淡黄色固体产物,其结构式见式23(产率为:97.3%)。
实例24
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的1l三口圆底烧瓶中,加入dmf(500ml,已除水)、产物1(57.82g,0.20mol),搅拌并升温至120℃。称取双酚a型环氧树脂e44(环氧值0.45,15.00g)、乙二胺(1.20g,0.02mol)加入烧杯中,加dmf(50ml,已除水)搅拌至完全溶解,加入到上述溶液。在120℃下反应8小时。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得71.13g包覆后的淡黄色固体产物,其结构式见式24(产率为:96.1%)。
实例25
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入dmf(200ml,已除水)、产物1(28.91g,0.10mol),搅拌并升温至120℃。称取双酚s型环氧树脂(环氧值0.38,10.00g)、乙二胺(0.60g,0.01mol)加入烧杯中,加dmf(30ml,已除水)搅拌至完全溶解,加入到上述溶液。在120℃下反应12小时。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得37.81g包覆后的淡黄色固体产物,其结构式见式25(产率为:95.7%)。
实例26
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的1l三口圆底烧瓶中,加入dmf(500ml,已除水)、产物1(57.82g,0.20mol),搅拌并升温至120℃。称取双酚f型环氧树脂(环氧值0.66,10.00g)、乙二胺(1.20g,0.02mol)加入烧杯中,加dmf(50ml,已除水)搅拌至完全溶解,加入到上述溶液。在120℃下反应12小时。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得75.95g包覆后的淡黄色固体产物,其结构式见式23(产率为:98.1%)。
实例27
在装有球形冷凝管、磁力加热搅拌,温度计及滴液漏斗的500ml三口圆底烧瓶中,加入dmf(200ml,已除水)、产物1(28.91g,0.20mol),搅拌并升温至140℃。称取酚醛型环氧树脂f-44(环氧值0.54,10.00g)、乙二胺(1.20g,0.02mol)加入烧杯中,加dmf(30ml,已除水)搅拌至完全溶解,加入到上述溶液。在140℃下反应8小时。反应液室温下冷却,并用无水乙醇洗三次,过滤并真空干燥得39.19g包覆后的淡黄色固体产物,其结构式见式23(产率为:97.7%)。
将本发明的阻燃剂用于聚丙烯中,与商品阻燃剂聚磷酸铵/季戊四醇(对比例)体系的性能比较如表1所示。
表1不同阻燃剂体系在聚丙烯中的阻燃性能
上述实施例并非是对于本发明的限制,本发明并非仅限于上述实施例,只要符合本发明要求,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!