一步合成氨基吡啶及4-氨基吡啶的方法与流程
本发明属于化工原料合成的技术领域,更具体地说,涉及一步合成氨基吡啶及4-氨基吡啶的方法。
背景技术
4-氨基吡啶是一种重要的农药、医药及染料中间体;也用作化学试剂。合成抗生素类药物4-乙酰胺基哌啶醋酸盐等的中间体,也是制备强心剂、灭菌剂、抗心律失常药、抗溃疡药及解痉药米尔维林的原料;新型降压药吡那地尔的重要原料。
目前制备4-氨基吡啶的方法有金属还原法、催化剂加氢法、电化学还原法等。目前,国内外产业生产中制备4-氨基吡啶绝大多数采用金属还原法,或醋酸体系来实现硝基的还原,收率达85%。国外采用的低价的金属钛浆状物或他的氯化物对硝基氮氧吡啶进行还原,产品收率在90%以上。虽然可得到高收率的产品,但催化剂的制备较难,需要在无水条件下进行,且对原料纯度要求较高,不利于工业化生产。国内外学者提出催化剂加氢4-硝基吡啶n.氧化物的方法,并取得了良好效果。常用的加氢催化剂有pto2,raney镍。主要是由于该类催化剂活性较高,选择性好且pto2,raneyni镍对氮氧键具有较好的还原能力。薛勇等研究了在hcoonh4-{4-pd/c条件下,在乙醇溶剂中于60度常压催化加氢制备4-氨基吡啶,收率为90%。赵铭等采用pd/alo3做催化剂,无水乙醇作溶解,催化合成了4-氨基吡啶,收率为85.6%。王志祥等人研究了以raneyni为催化剂,95%乙醇中于室温常压下通入氮气,滤去催化剂,蒸去乙醇得暗红色固体,用苯-石油醚重结晶,得白色针状晶体4-氨基吡啶,收率为89%。作为“绿色化学”过程中重要分支的电化学引起了人们的关注。jhranilovic等曾采用汞作阴极,铂丝作阳极,在酸性体系中对吡啶类氮氧化物的电化学还原过程进行研究,4-氨基吡啶的收率为59.5%,电流效率为30~35%。
例如,中国专利申请号为201210383381.7,申请日为2012年10月11日的专利申请文件公开了一种氨基吡啶的合成方法,在卤代吡啶和液氨的混合液中加入溶剂和催化剂,于200-1000℃温度下反应得到粗品,再由甲苯精制得到目标产品,所述催化剂为负载量为10%的钌碳催化剂,所述溶剂为喹啉。中国专利申请号为201610609618.7,申请日为2016年7月28日的专利申请文件公开了一种合成4-氨基吡啶的方法,采用了4-氯吡啶为起始原料,在催化剂作用下,与液氨在压力为0.8~3.0mpa,温度为30~90℃下在反应4~15小时后,去除氨,经水洗、过滤,烘干得到目标产品4-氨基吡啶,所采用的催化剂为氯化铁、氯化锌、氯化铜中的一种,催化剂的使用量为4-氯吡啶的1~10%。目前公开的氨基吡啶的合成方法都存在反应条件要求高、三废污染多、需要多步反应导致收率低等问题。
近年来国家对环保问题非常重视,多次启动中央环境保护督察组进驻到全国各地督查企业的环保问题,特别是化工类生产企业,是环保督查的重点对象,现有技术中公开的氨基吡啶合成方法,后期三废处理的成本太高,而且资源的浪费比较严重,因此不适合工业化生产。但是氨基吡啶又是一种重要的化工原料,市场需求量比较大,因此研发一种环保、高效、经济的氨基吡啶制备方法意义重大。
技术实现要素:
1.要解决的问题
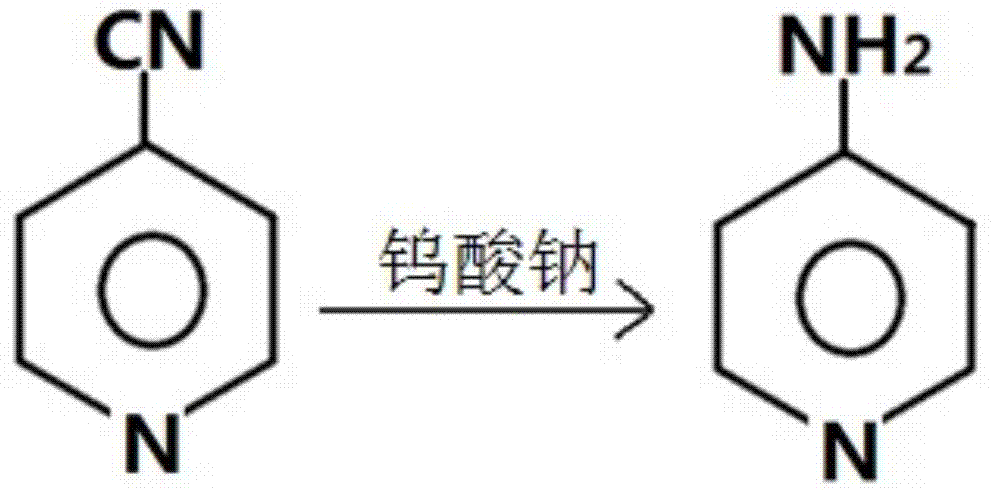
针对现有氨基吡啶制备方法存在条件要求高、步骤繁琐、三废污染多、产率低下等问题,本发明提供一步合成氨基吡啶及4-氨基吡啶的方法,本发明利用钨酸钠作为催化剂,在次氯酸钠水溶剂条件即可将氰基吡啶反应一步生成氨基吡啶,特别是4-氰基吡啶反应生成4-氨基吡啶,然后用氯仿进行萃取,蒸馏脱除溶剂后,得到氨基吡啶产品,产品的纯度能高达99.5%左右,反应的产率由现有技术中的50%多提高到85%。
2.技术方案
为了解决上述问题,本发明所采用的技术方案如下:
一步合成氨基吡啶的方法,以氰基吡啶为原料,以钨酸钠作为催化剂,一步反应得到。
更进一步地,反应溶剂为次氯酸钠水溶液。
一步合成4-氨基吡啶的方法,以4-氰基吡啶为原料,以钨酸钠作为催化剂,在次氯酸钠水溶液中一步反应得到。
更进一步地,包括以下步骤:
(1)制备4-氰基吡啶水溶液,降温至0℃以下;
(2)加入催化剂,加入量为4-氰基吡啶质量的2~5%;
(3)滴加次氯酸钠溶液至反应液中反应,中控分析,使反应液中的4-氰基吡啶含量小于0.8%;
(4)将步骤(3)中的反应液滴加入次氯酸钠水溶液中,升温至90~95℃保持10小时以上,反应结束;
(5)反应结束后,对反应液进行萃取和蒸馏,得到4-氨基吡啶。
更进一步地,制备的4-氰基吡啶水溶液的质量浓度为20~30%。
更进一步地,催化剂为钨酸钠。
更进一步地,步骤(3)中的次氯酸钠水溶液质量浓度为10~11%,滴加的次氯酸钠水溶液的质量为4-氰基吡啶质量的2.5~3.5倍。
更进一步地,步骤(4)中的次氯酸钠水溶液质量浓度为8-12%,次氯酸钠水溶液的质量为4-氰基吡啶质量的3~4倍。
更进一步地,反应结束后,用氯仿对反应液进行萃取,至少萃取3次,合并氯仿层,在常压下蒸馏,回收氯仿,然后再减压蒸馏得到4-氨基吡啶。
3.有益效果
相比于现有技术,本发明的有益效果为:
(1)本发明提供的一步合成氨基吡啶及4-氨基吡啶的方法,在低温条件下,以钨酸钠作为催化剂,在次氯酸钠的水溶液中,4-氰基吡啶先反应停留在4-甲酰胺吡啶上,再升温进行霍夫曼降解得到4-氨基吡啶;如果没有钨酸钠,则在次氯酸钠的作用下,4-氰基吡啶会先反应到4-甲酸吡啶,而钨酸钠催化剂能抑制4-氰基吡啶反应到4-甲酸吡啶,促进4-氰基吡啶反应到4-甲酰胺吡啶,然后再升温条件下一步反应得到4-氨基吡啶;现有技术中通常将4-氰基吡啶在碳酸钠的作用下,先生成4-甲酰胺吡啶,再投入次氯酸钠,得到4-氨基吡啶,这种传统方法的缺陷是两次使用不同的碱性盐,碳酸钠用量大,碱性弱,产生高盐分的废水,增加废水的量,而且碳酸钠易生成二氧化碳易起泡沫,不利于反应的进行,产率低;
(2)本发明提供的4-氨基吡啶的制备方法,条件易于控制,反应步骤简单,产物分离方便,副反应少,产率能达到85%,产品纯度能达到99.5%;
(3)本发明提供的制备4-氨基吡啶的方法,巧妙的两次利用次氯酸钠溶液,分别利用次氯酸钠的不同性质,第一次利用次氯酸钠的碱性,水解到4-甲酰胺吡啶;第二次利用次氯酸钠的氧化性,进行霍夫曼降解,这其中,还创新性的使用钨酸钠作为催化剂,控制反应的进行,抑制了副反应的发生,所以本发明的反应副产物少(酸类产物),产率高。
附图说明
图1为本发明的合成流程示意图;
图2为本发明中产品的气相色谱图。
具体实施方式
下面结合具体实施例对本发明进一步进行描述。
实施例1
如图1所示,本发明提供了一种一步合成氨基吡啶的方法,主要的步骤包括:在低温条件下,以氰基吡啶为原料,以钨酸钠作为催化剂,在次氯酸钠水溶液中先预反应,然后升温进行霍夫曼一步反应得到,本发明摸索得到的这种制备方法与现有技术相比,具有反应条件易控制、副产物少、三废污染少等优点,更适用于工业应用。以4-氨基吡啶为例,采用上述方法进行合成,主要步骤为:
(1)制备4-氰基吡啶水溶液(在反应瓶中,用375g水溶解100g4-氰基吡啶,得到4-氰基吡啶水溶液),将水溶液降温至0℃以下;
(2)加入催化剂钨酸钠,加入量为4-氰基吡啶质量的5%,即加入5g催化剂;
(3)然后慢慢滴加次氯酸钠溶液(次氯酸钠溶液的质量浓度为10%,加入量为4-氰基吡啶质量的3倍,即加入了300g10%浓度的次氯酸钠溶液)至反应液中反应,中控分析,使反应液中的4-氰基吡啶含量小于0.8%;
(4)将步骤(3)中的反应液滴加入到预先制备好的次氯酸钠水溶液(次氯酸钠溶液的质量浓度为12%,加入量为4-氰基吡啶质量的4倍,即,将反应液加入了到400g12%浓度的次氯酸钠溶液)中,升温至95℃保持10小时以上进行反应,本实施例中保持了10小时左右,反应结束;
(5)反应结束后,对用氯仿对反应液进行萃取,至少萃取3次,本次实施例中选择萃取3次,然后合并氯仿层进行常压蒸馏,回收氯仿溶液用于下批反应液的萃取,常压蒸馏之后,在进行减压蒸馏,蒸出4-氨基吡啶产品,产品外观为白色晶体状。
对反应的产物4-氨基吡啶进行分析,分析仪器为:灵华gc9890b;色谱柱型:se-5430m*0.32mm*0.4μm;检测的仪器条件:柱温:140℃检测器:280℃汽化温:280℃;灵敏度:3。
样品处理:
将送检样品取样0.5g,加3ml甲醇完全后进样。用10ul进样器取1格0.2ul进样,140程序升温(140℃保持1min,20℃/min程序升温至280℃保持5min)锁掉溶剂峰,面积归一法测其含量,色谱图如图2所示,图中各峰的具体信息见下表,产品的纯度高达99.89%,纯度非常高,制备方法相对现有技术方便、经济,三废污染小,副反应少,得到的产品纯度高,同时产率也非常高。
表14-氨基吡啶产品的色谱图分析结果表
实施例2
如图1所示,本发明提供的一步合成氨基吡啶的方法,主要的步骤包括:在低温条件下,以氰基吡啶为原料,以钨酸钠作为催化剂(催化剂的加入量为原料氰基吡啶质量的2~5%左右),在次氯酸钠水溶液中先预反应,然后升温进行霍夫曼一步反应得到。以4-氨基吡啶为例,采用上述方法进行合成,主要步骤为:
(1)制备4-氰基吡啶水溶液(在反应瓶中,用400g水溶解171g4-氰基吡啶,得到4-氰基吡啶水溶液),将水溶液降温至0℃以下;
(2)加入催化剂钨酸钠,加入量为4-氰基吡啶质量的2%,即加入3.4g催化剂;
(3)然后慢慢滴加次氯酸钠溶液(次氯酸钠溶液的质量浓度为11%,加入量为4-氰基吡啶质量的2.5倍,即加入了428g11%浓度的次氯酸钠溶液)至反应液中反应,中控分析,使反应液中的4-氰基吡啶含量小于0.8%;
(4)将步骤(3)中的反应液滴加入到预先制备好的次氯酸钠水溶液(次氯酸钠溶液的质量浓度为10%,加入量为4-氰基吡啶质量的3倍,即,将反应液加入了到513g10%浓度的次氯酸钠溶液)中,升温至95℃保持10小时以上进行反应,本实施例中保持了12小时左右,反应结束;
(5)反应结束后,对用氯仿对反应液进行萃取,至少萃取3次,本次实施例中选择萃取4次,然后合并氯仿层进行常压蒸馏,回收氯仿溶液用于下批反应液的萃取,常压蒸馏之后,在进行减压蒸馏,蒸出4-氨基吡啶产品,产品外观为白色晶体状。
对反应的产物4-氨基吡啶进行分析,分析仪器为:灵华gc9890b;色谱柱型:se-5430m*0.32mm*0.4μm;检测的仪器条件:柱温:140℃检测器:280℃汽化温:280℃;灵敏度:3。
样品处理:
将送检样品取样0.5g,加3ml甲醇完全后进样。用10ul进样器取1格0.2ul进样,140程序升温(140℃保持1min,20℃/min程序升温至280℃保持5min)锁掉溶剂峰,面积归一法测其含量,产品的纯度高达99.91%,纯度非常高,制备方法相对现有技术方便、经济,三废污染小,副反应少,得到的产品纯度高,同时产率也高达85%。
实施例3
如图1所示,本发明提供的一种一步合成氨基吡啶的方法,主要的步骤包括:在低温条件下,以氰基吡啶为原料,以钨酸钠作为催化剂(催化剂的加入量为原料氰基吡啶质量的2~5%左右),在次氯酸钠水溶液中先预反应,然后升温进行霍夫曼一步反应得到。以4-氨基吡啶为例,采用上述方法进行合成,主要步骤为:
(1)制备4-氰基吡啶水溶液(在反应瓶中,用300g水溶解100g4-氰基吡啶,得到4-氰基吡啶水溶液),将水溶液降温至0℃以下;
(2)加入催化剂钨酸钠,加入量为4-氰基吡啶质量的3%,即加入3g催化剂;
(3)然后慢慢滴加次氯酸钠溶液(次氯酸钠溶液的质量浓度为10%,加入量为4-氰基吡啶质量的3.5倍,即加入了350g10%浓度的次氯酸钠溶液)至反应液中反应,中控分析,使反应液中的4-氰基吡啶含量小于0.8%;
(4)将步骤(3)中的反应液滴加入到预先制备好的次氯酸钠水溶液(次氯酸钠溶液的质量浓度为8%,加入量为4-氰基吡啶质量的3.5倍,即,将反应液加入了到350g8%浓度的次氯酸钠溶液)中,升温至90℃保持10小时以上进行反应,本实施例中保持了15小时左右,反应结束;
(5)反应结束后,对用氯仿对反应液进行萃取,至少萃取3次,本次实施例中选择萃取3次,然后合并氯仿层进行常压蒸馏,回收氯仿溶液用于下批反应液的萃取,常压蒸馏之后,在进行减压蒸馏,蒸出4-氨基吡啶产品,产品外观为白色晶体状。
对反应的产物4-氨基吡啶进行分析,分析仪器为:灵华gc9890b;色谱柱型:se-5430m*0.32mm*0.4μm;检测的仪器条件:柱温:140℃检测器:280℃汽化温:280℃;灵敏度:3。
样品处理:
将送检样品取样0.5g,加3ml甲醇完全后进样。用10ul进样器取1格0.2ul进样,140程序升温(140℃保持1min,20℃/min程序升温至280℃保持5min)锁掉溶剂峰,面积归一法测其含量,产品的纯度高达99.95%,纯度非常高,制备方法相对现有技术方便、经济,三废污染小,副反应少,得到的产品纯度高,同时产率也高达85%。
实施例4
本发明提供的一种一步合成氨基吡啶的方法,主要的步骤包括:在低温条件下,以氰基吡啶为原料,以钨酸钠作为催化剂(催化剂的加入量为原料氰基吡啶质量的2~5%左右),在次氯酸钠水溶液中先预反应,然后升温进行霍夫曼一步反应得到。以4-氨基吡啶为例,采用上述方法进行合成,主要步骤为:
(1)制备4-氰基吡啶水溶液(在反应瓶中,用400g水溶解100g4-氰基吡啶,得到4-氰基吡啶水溶液),将水溶液降温至0℃以下;
(2)加入催化剂钨酸钠,加入量为4-氰基吡啶质量的4%,即加入4g催化剂;
(3)然后慢慢滴加次氯酸钠溶液(次氯酸钠溶液的质量浓度为11%,加入量为4-氰基吡啶质量的3倍,即加入了300g11%浓度的次氯酸钠溶液)至反应液中反应,中控分析,使反应液中的4-氰基吡啶含量小于0.8%;
(4)将步骤(3)中的反应液滴加入到预先制备好的次氯酸钠水溶液(次氯酸钠溶液的质量浓度为9%,加入量为4-氰基吡啶质量的4倍,即,将反应液加入了到400g9%浓度的次氯酸钠溶液)中,升温至95℃保持10小时以上进行反应,本实施例中保持了14小时,反应结束;
(5)反应结束后,对用氯仿对反应液进行萃取,至少萃取3次,本次实施例中选择萃取3次,然后合并氯仿层进行常压蒸馏,回收氯仿溶液用于下批反应液的萃取,常压蒸馏之后,在进行减压蒸馏,蒸出4-氨基吡啶产品,产品外观为白色晶体状。
对反应的产物4-氨基吡啶进行分析,分析仪器为:灵华gc9890b;色谱柱型:se-5430m*0.32mm*0.4μm;检测的仪器条件:柱温:140℃检测器:280℃汽化温:280℃;灵敏度:3。
样品处理:
将送检样品取样0.5g,加3ml甲醇完全后进样。用10ul进样器取1格0.2ul进样,140程序升温(140℃保持1min,20℃/min程序升温至280℃保持5min)锁掉溶剂峰,面积归一法测其含量,产品的纯度高达99.85%,纯度非常高,制备方法相对现有技术方便、经济,三废污染小,副反应少,得到的产品纯度高,同时产率也高达85%。
对比例1
以4-氨基吡啶为例,在不使用催化剂的情况下,采用类似的上述方法进行合成,主要步骤为:
(1)制备4-氰基吡啶水溶液(在反应瓶中,用380g水溶解100g4-氰基吡啶,得到4-氰基吡啶水溶液),将水溶液降温至0℃以下;
(2)然后慢慢滴加次氯酸钠溶液(次氯酸钠溶液的质量浓度为10%,加入量为4-氰基吡啶质量的3倍,即加入了300g10%浓度的次氯酸钠溶液)至反应液中反应,中控分析,使反应液中的4-氰基吡啶含量小于0.8%;检测分析发现有较多副产物生成,主要是4-吡啶甲酸,因此后续升温进行霍夫曼降解时,得到的4-氨基吡啶纯度低,且产率低,不到50%。
- 还没有人留言评论。精彩留言会获得点赞!