一种水溶性聚轮烷交联剂及其制备方法与流程
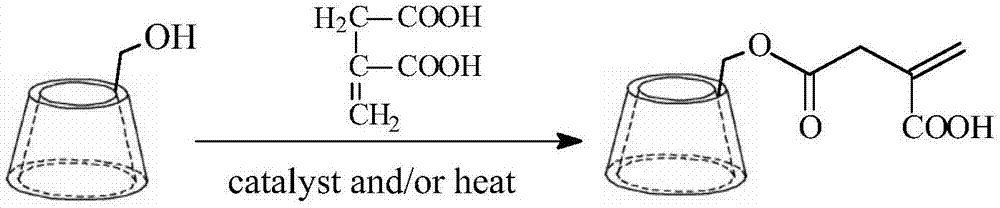
本发明属于交联剂制备技术领域,具体涉及一种水溶性聚轮烷交联剂及其制备方法。
背景技术
水凝胶是一种经适度交联从而具有三维网络结构的高分子材料,因其独特的吸水性、保水性及仿生特性,被广泛应用于工业、农业、医药和生物工程材料等领域。
目前水凝胶的合成大都是以传统的化学交联剂进行交联,传统的化学交联剂指的是一个分子式内具有两个或两个以上的双键,双键参与聚合在形成网络结构的过程中,由于交联过程具有随机性,不可避免地导致交联点在凝胶空间中分布不均匀,使得各交联点间的聚合物链段长短不一。而化学交联点又是固定的,因此在外力作用下聚合物链段受力不均匀,短链段受力大,易先断裂,导致整个网络结构遭到破坏。这就导致制备出的凝胶脆性大,形变能力差。
zl201710929543.5公开了一种通用型凝胶类聚轮烷交联剂及其制备方法,该交联剂可以与可聚合单体交联制备凝胶,其交联点不是固定的,交联点可沿聚合物链滑动,产生“滑动效应”使得外力被均匀分散到各个链段进而被分散到整个网络,从而较好解决化学交联点在空间分布不均匀的问题。这种聚轮烷交联剂易溶于有机溶剂,较适用于制备油凝胶,但是在水中的溶解度有限,经试验测定仅有0.6713g/100g水(20℃),属于微溶物质,限制了其在水溶液中实施水溶性单体交联制备水凝胶的应用。
技术实现要素:
为解决以上技术问题,本发明的目的在于提供一种水溶性聚轮烷交联剂及其制备方法。
为实现上述发明目的,具体提供了如下所述的技术方案:
1、一种水溶性聚轮烷交联剂的制备方法,包括如下步骤:
1):将α-环糊精进行衣康酸酐改性引入双键和羧酸结构,得改性α-环糊精;
2):将聚乙二醇两端的羟基氧化为羧酸,得改性聚乙二醇链;
3):将改性α-环糊精贯穿于第二步制备的改性聚乙二醇链上,再将改性聚乙二醇链两端封端,制备出水溶性聚轮烷交联剂。
进一步,步骤1)所述改性α-环糊精按如下方法进行制备:
步骤a:将α-环糊精、衣康酸溶解在装有无水乙醇的反应器中,然后将反应器置于干燥箱中进行干燥;
步骤b:将反应器取出并加热至反应物呈粘稠状黄色液体少许,再将反应器置于干燥器中将液体蒸干;
步骤c:进一步向反应器中加入无水乙醇并搅拌均匀,使用0.45um微孔滤膜进行抽虑得固体,将抽滤后的固体进行干燥得浅黄色粉末状固体。
更进一步,步骤1)所述改性α-环糊精按如下方法进行制备:
步骤a:将1.9~2gα-环糊精、0.1~0.2g衣康酸溶解在装有250ml无水乙醇的反应器中,然后将反应器置于干燥箱中在110℃条件下干燥130min;
步骤b:将反应器取出并加热至沸,持续沸腾30min后,反应器中反应物呈粘稠状黄色液体少许,再将反应器置于干燥器中将液体蒸干;
步骤c:进一步向反应器中加入50ml无水乙醇并搅拌均匀,使用0.45um微孔滤膜进行抽虑得固体,再用无水乙醇洗涤,将抽滤后的固体在100℃温度条件下进行干燥9h,得浅黄色粉末状固体。
进一步,步骤2)所述氧化方法为:将聚乙二醇、四甲基哌啶氮氧化物、nabr和naclo依次加入蒸馏水中,调节ph值为10-11,常温下反应10-15min,然后加入乙醇停止氧化,并调节ph<2后用ch2cl2萃取,取下层清液,将该下层清液真空干燥即得所述改性聚乙二醇链。
更进一步的,步骤2)还包括重结晶,真空干燥后所得的改性聚乙二醇链溶解在50-60℃的乙醇中重结晶,再次真空干燥即可。
优选的,所述聚乙二醇、四甲基哌啶氮氧化物和nabr的质量比为100:1:1;所述nabr与naclo的质量体积比(g/ml)为10:1。
进一步,步骤3)所述贯穿并封端的步骤为:称取改性α-环糊精和改性聚乙二醇链溶解在蒸馏水中,冷藏过夜得白色络合物,将该白色络合物与1-金刚烷胺、卡特缩合剂、n,n-二异丙基乙胺一起加至n,n-二甲基酰胺中并混合均匀呈白色浆液,将该白色浆液在0~10℃下过夜得乳白色浆液,然后将乳白色浆液与甲醇混合离心洗涤两次后,再用蒸馏水反复洗涤,抽滤并真空干燥得到水溶性聚轮烷交联剂。
优选的,改性α-环糊精和改性聚乙二醇链称取质量比为3.8~4.2:1。
进一步优选的,所述白色络合物与1-金刚烷胺、卡特缩合剂、n,n-二异丙基乙胺、n,n-二甲基酰胺的添加质量比例(g:g:g:ml:ml)为:0.5~0.7:0.01~0.02:0.04~0.05:0.01~0.03:9~12。
2、由所述制备方法得到的水溶性聚轮烷交联剂。
本发明的有益效果在于:本发明公开了一种水溶性聚轮烷交联剂的制备方法,通过将α-环糊精进行衣康酸酐改性引入双键和羧酸结构结构;将聚乙二醇两端的羟基氧化为羧酸;将制备的改性环糊精贯穿于第二步制备的改性聚乙二醇链上,再将改性聚乙二醇链两端封端,制备出聚轮烷交联剂。
由于α-环糊精改性中引入了羧酸基团,增强了聚轮烷的水溶性,因此可以和丙烯酰胺、丙烯酸等水溶性单体交联聚合制备水凝胶,提高水凝胶的形变能力。
附图说明
图1为α-环糊精的改性过程示意图;
图2为聚乙二醇的氧化过程示意图;
图3为水溶性聚轮烷交联剂的制备过程示意图;
图4为衣康酸、α-环糊精、改性α-环糊精的核磁共振h谱图;
图5为聚乙二醇、氧化聚乙二醇的核磁共振h谱图;
图6为水溶性聚轮烷交联剂的红外光谱图;
图7为水溶性聚轮烷交联剂的核磁共振h谱;
图8为水溶性聚轮烷交联剂的x射线衍射谱图;
图9为传统交联剂制备凝胶受力前后形态照片;
图10为水溶性聚轮烷交联剂制备凝胶受力前后形态照片。
具体实施方式
下面将结合具体实施例及附图对本发明进行更加详细的阐述。
以下实施例所需原材料及简称如下:α-环糊精(α-cd),聚乙二醇(peg),次氯酸钠(naclo),丙烯酰胺,二甲基亚砜(dmso),四甲基哌啶氮氧化物(tempo),1-金刚烷胺,n,n-二甲基酰胺(dmf),n,n-二异丙基乙胺(edipa),卡特缩合剂(bop试剂)等。
实施例
一、制备试验样品
一种水溶性聚轮烷交联剂的制备:
1)改性α-环糊精的制备:在烧杯中加入250ml无水乙醇,再加入1.9909gα-cd和0.1348g衣康酸,搅拌30min,完全溶解后放入110℃的干燥箱中130min,再将烧杯取出,使用加热至沸,持续沸腾30min后,烧杯中呈粘稠状黄色液体少许。再将烧杯放入干燥箱中将液体蒸干。倒入50ml无水乙醇,使用0.45um微孔滤膜进行抽虑,再用少量无水乙醇洗涤,将抽滤后的固体放入100℃的干燥箱内干燥9h,得到浅黄色粉末状固体,即为改性α-环糊精(简称改性α-cd),其反应过程如图1所示;
2)聚乙二醇的氧化:在蒸馏水中加入0.1002gtempo、0.1013gnabr充分搅拌至两种物质完全溶解,向其中加入10.0468gpeg,10mlnaclo搅拌,室温下反应15min。再加入10ml乙醇停止氧化,用hcl调节至ph<2,并用ch2cl2萃取三次,取下层清液合并后溶解于125ml热乙醇中,放入冰箱过夜。取出40℃真空干燥20h,得到白色的聚乙二醇链,其反应过程如图2所示;
3)改性α-环糊精贯穿于聚乙二醇链,并封端:取0.6002g改性α-cd和0.1503g氧化peg溶于10ml蒸馏水中,溶解后为白色液体。将溶解的混合液放入冰箱中保持过夜,得到0.6931g白色络合物。将络合物与0.0166g1-金刚烷胺、0.0482g卡特缩合剂(bop试剂)、0.02mln,n-二异丙基乙胺(edipa)溶解在10mldmf中,观察到混合液为白色浆液。混合浆液0~10℃下过夜,过夜后拿出冰箱混合液为乳白色浆液,随后用甲醇抽滤洗涤两次,真空干燥得到0.1415g的白色聚轮烷交联剂,即水溶性聚轮烷交联剂,起反应过程如图3所示。
二、原料、中间物及产物的表征
取α-环糊精、衣康酸、改性α-环糊精、聚乙二醇、氧化后的聚乙二醇链以及经过上述试验例制备的水溶性聚轮烷交联剂进行核磁共振h谱、红外光谱等表征。
1)改性α-cd的表征
使用氘代dmso做溶剂使用核磁共振手段表征衣康酸、α-环糊精、改性α-环糊精,结果如图4所示。
图4中谱线a为衣康酸的谱图,3.15ppm处为衣康酸中亚甲基氢原子的化学位移,5.63ppm和6.11ppm处分别为衣康酸双键碳原子相连的两个氢原子的化学位移。谱线b为α-环糊精的谱图。谱线c为改性α-环糊精的谱图,该谱图在3.15、5.63和6.11ppm处也出现了氢原子的化学位移峰,表明改性α-环糊精结构中存在衣康酸的化学结构,即改性成功。
2)氧化后的聚乙二醇链的表征:
使用氘代dmso做溶剂使用核磁共振手段表征聚乙二醇、氧化后的聚乙二醇链,结果如图5所示:
图5中谱线a为聚乙二醇的谱图,2.51ppm处为聚乙二醇中醇羟基氢原子的化学位移,3.41-3.56ppm处聚乙二醇中亚甲基氢原子的化学位移。谱线b为氧化后的聚乙二醇链的谱图,对比谱线a,3.98ppm处新出现了氢原子峰,为氧化后与羧酸基团直接相连的亚甲基氢原子的化学位移,而2.51ppm处的峰消失了,表明氧化聚乙二醇链中不存在醇羟基氢原子,即聚乙二醇的端羟基全部氧化为羧酸。
3)水溶性聚轮烷交联剂的表征
红外光谱表征:由上述实施例制备所得水溶性聚轮烷交联剂的红外光谱图,如图6所示,图中3400cm-1位置处的宽吸收峰可能是胺基n-h键或者酸羟基o-h键的伸缩振动峰,2931cm-1位置处可能是与双键碳相连的c-h键的振动吸收峰,1638cm-1位置处可能为c=c双键的伸缩振动峰,1148cm-1位置处可能为c-o-c键的不对称伸缩振动峰,由此可见交联剂中主要的特征基团在红外谱图中均存在吸收峰。
核磁共振h谱表征:以重水做溶剂使用核磁共振手段表征水溶性聚轮烷交联剂,如图7所示,3.81-3.89ppm、3.66-3.80ppm和3.41-3.52ppm处均为环糊精分子上来自不同位置的氢原子化学位移,但是3.54-3.61ppm处应为聚乙二醇结构中亚甲基氢原子的化学位移,该交联剂中既存在环糊精结构又存在聚乙二醇结构。需要说明的是图7中环糊精分子中氢原子化学位移和聚乙二醇结构中亚甲基氢原子的化学位移与图4、图5相比会有一些偏移,因为使用了不同的溶剂。
x射线衍射表征:表征结果如图8所示,在在20°左右出现了明显的衍射信号,表明改性环糊精和改性聚乙二醇形成了包合物,晶体堆砌类型为隧道状结构。
三、水溶性测试
将本实施例制备的水溶性聚轮烷交联剂与zl201710929543.5公开的通用型凝胶类聚轮烷交联剂进行水溶性测试比较,得到如表1所示的结果:
表1水溶性测试试验数据
由表1测试数据可显著说明,本发明制备的聚轮烷交联剂在水中的溶解度相比于通用型凝胶类聚轮烷交联剂具有非常显著的提高。
四、水溶性聚轮烷交联剂、通用型聚轮烷交联剂用于制备凝胶的应用及性能比对:
在两个烧杯中分别加入70g蒸馏水和14g丙烯酰胺,再各加入占单体质量0.2%的v50,分别加入传统交联剂n,n′-亚甲基双丙烯酰胺和新型交联剂各占单体质量的0.5%,70℃下反应4h,得到的产物同时在70℃下真空干燥16h。
取大小相同的长、宽、高分别为2.1cm、1.5cm、0.6cm的两种体膨颗粒,分别放入装有500ml蒸馏水烧杯中,吸水10h后,分别取相同大小的凝胶。将传统凝胶中心对准nk系列推拉力计的中心轴处,调零,逆时针旋转拉力计上方旋钮使中心轴向下挤压,凝胶受力,待传统凝胶破碎后记录表盘示数。在相同条件下重复上述步骤测量新型凝胶的形变性能。如图9所示,传统交联剂合成的凝胶受到174.2n的力时,凝胶破碎。如图10所示,新型交联剂合成的凝胶在受到286.4n的力后,再撤去外力后,凝胶并未破碎且能恢复至原始状态,以上可说明水溶性聚轮烷交联剂具有滑动效应,因此形变能力和恢复能力强,传统交联剂n,n′-亚甲基双丙烯酰胺没有滑动效应,因此形变能力和恢复能力差。
最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!