依达拉奉衍生物及其制备方法、检测方法和用途与流程
发明领域本发明涉及一种依达拉奉衍生物,即(z)-2-(3-甲基-5-氧-1-苯基-4,5-二氢-1h-吡唑-4-基)-3-[(e)-苯偶氮基]-2-丁烯酸,及其制备方法、检测方法和用途。
背景技术:
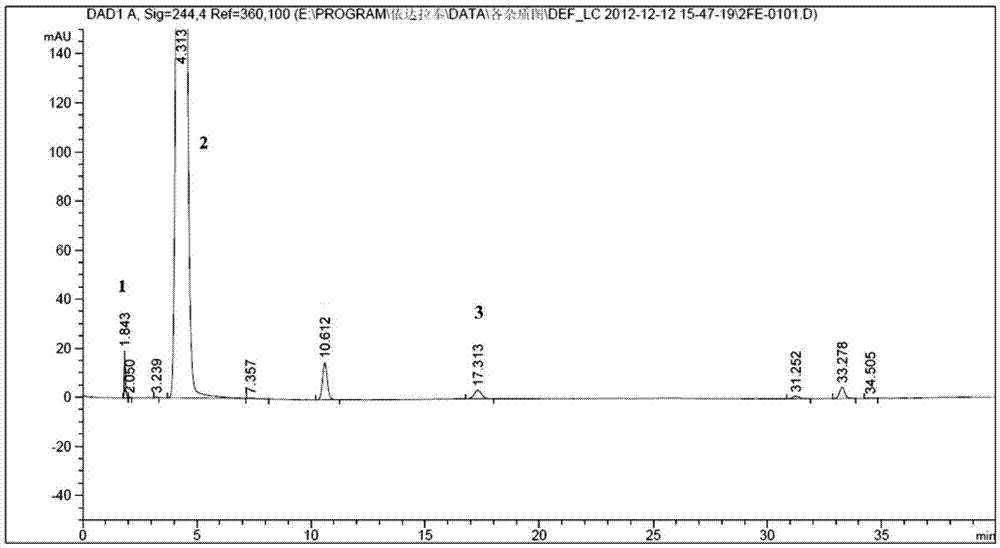
:依达拉奉(式i化合物)是一种脑保护剂。脑梗塞急性期患者给予依达拉奉,可抑制梗塞周围局部脑血流量的减少,使发病后第28天脑中naa含量较甘油对照组明显升高。依达拉奉可清除自由基,抑制脂质过氧化,从而抑制脑细胞、血管内皮细胞、神经细胞的氧化损伤。作为临床常用药物,依达拉奉在其生产、贮存、使用过程中由于灭菌等高温过程的存在,有可能产生杂质而导致药物质量下降。国内对于依达拉奉在生产、贮存等过程中的相关物质研究的较多,比如反应原料乙酰乙酸乙醋和苯肼,在依达拉奉注射液的制备及长期存放中产生的3,3'-二甲基-1,1'-二苯基-1h,1'h-[4,4'-二吡唑]-5,5'(4h,4'h)-二酮等。对于依达拉奉原料药在存放过程中产生的有关物质报道较少。因此,对依达拉奉原料在制备和存放过程中产生的杂质及杂质制备方法、检测方法进行研究是十分有必要的。技术实现要素:我们经研究发现,由于氧化反应等,依达拉奉原料在存放过程中会产生杂质,我们对放置一定时间的依达拉奉原料采用高效液相色谱进行分离检测,发现确有杂质峰,其中包括一种新的杂质化合物。具体方法为将上述依达拉奉原料用甲醇:水:冰醋酸=45:55:0.1的混合溶液溶解并定容,配置成依达拉奉含量为0.2mg/ml的样品,然后采用高效液相色谱法,色谱柱为agilenteclipsexdb-c18,4.6*150mm(3.5μm),流动相a为甲醇:水:冰醋酸=45:55:0.1,流动相b为甲醇,流动相流速为0.8ml/min,柱温为20℃,检测波长为244nm,进样量为20ml。梯度洗脱如下:时间(min)流动相a比例(%)流动相b比例(%)01000221000523664723664801000检测结果发现出峰时间为17.3min的化合物是一种新的化合物。本发明的目的之一即是提供此种如下式ⅱ结构的新化合物:(z)-2-(3-甲基-5-氧-1-苯基-4,5-二氢-1h-吡唑-4-基)-3-[(e)-苯偶氮基]-2-丁烯酸,以下简称化合物a。在一定条件下,化合物a的烯醇式和酮式可以相互转化。本发明的目的之二是提供一种制备上述化合物a的方法,为实现上述目的,采用以下制备分离工艺:将依达拉奉加入溶剂配制成溶液或浑浊液,再与过氧化氢水溶液进行反应后,旋去部分溶剂,用反相高效液相色谱进行分离,0-40℃下避光旋干后即得。进一步,依达拉奉和过氧化氢的质量比为1:1~1:3,优选1:2~1:2.5。再进一步,所述溶剂为有机溶剂,所述有机溶剂选自醇类、乙腈,醇类优选甲醇,所述依达拉奉与过氧化氢水溶液的反应温度为15~20℃,反应时间为48~72小时。再进一步,所述反相高效液相色谱采用十八烷基硅烷键合硅胶为填充剂的色谱柱,流动相是有机相和水相的混合溶剂,检测波长为240~254nm,优选244nm。再进一步,流动相中有机相和水相的体积比为45:55~80:20,所述有机相选自甲醇或乙腈,所述水相ph为3.0~5.5,水相选自易挥发性弱酸水溶液,优选乙酸水溶液或甲酸水溶液。最终将化合物a用hplc分析其纯度,并采用ms、nmr确证其结构。本发明的目的之三是提供一种检测上述化合物a的方法。该方法为采用高效液相色谱,以十八烷基硅烷键合硅胶为填充剂,流动相为有机相和水相的混合溶剂,流动相流速为0.7~0.9ml/min,检测波长为240~254nm,其中所述流动相中有机相与水相的体积比为45:55~80:20。进一步,所述流动相选自甲醇水体系、甲醇缓冲盐体系、乙腈缓冲盐体系,优选甲醇-乙酸水溶液,流动相流速为0.8ml/min,检测波长为244nm。依达拉奉为全合成脑卒中神经保护剂,制剂及存放过程中由于存在高温灭菌、氧化等因素,其原料药或制剂中可能产生化合物a而导致质量下降。本发明采用的化合物a的制备方法简单,产品纯度高,检测方法简便,可作为依达拉奉杂质对照品,便于控制依达拉奉原料药及其相关制剂中化合物a的量。附图说明图1为依达拉奉、原料及杂质定位图。(其中色谱峰:1:苯肼;2:依达拉奉;3:(z)-2-(3-甲基-5-氧-1-苯基-4,5-二氢-1h-吡唑-4-基)-3-[(e)-苯偶氮基]-2-丁烯酸。)图2为依达拉奉及杂质定位图。(其中色谱峰a为(z)-2-(3-甲基-5-氧-1-苯基-4,5-二氢-1h-吡唑-4-基)-3-[(e)-苯偶氮基]-2-丁烯酸。)具体实施方式下面结合具体实施例对本发明作进一步阐述,但不限制本发明。依达拉奉原料药由吉林博大制药有限责任公司提供,其他所用试剂均为市售可得。实施例1:化合物a的制备取依达拉奉原料药10g加入甲醇200ml溶解,缓慢加入质量分数为30%的过氧化氢50ml,室温20℃搅拌48小时,10℃温度下旋去部分溶剂,溶液备用。然后采用反相高效液相色谱仪对上述溶液进行分离,色谱条件为:色谱柱为watersnova-pakc18,6μm,190*300mm,流动相a为甲醇:水:冰醋酸=45:55:0.1,流动相b为甲醇,流速:20ml/min,柱温:20℃,检测波长:244nm,进样量:0.5ml。梯度洗脱如下:时间(min)流动相a比例(%)流动相b比例(%)01000221000523664723664801000采集相对保留时间为3.0-3.6的制备液,10℃温度下旋干溶剂,得到化合物a5mg,收率0.048%。最终产品用核磁、质谱进行结构鉴定,附数据说明如下。化合物a:1、lc-ms(esi(+))分子离子峰(m+1),m/z=363.1;lc-ms(esi(-))分子离子峰(m-1),m/z=361.2;ms-ms碎片峰(esi(+)),m/z=345.2;m/z=317.1;m/z=77.1;(esi(-)),m/z=317.1;m/z=276.2。2、nmr(cd3od)化合物a在氘代甲醇中大部分以烯醇式形式存在:实施例2:化合物a的制备取依达拉奉原料药10g加入乙腈200ml溶解,缓慢加入质量分数为30%的过氧化氢50ml,室温20℃搅拌48小时,10℃温度下旋去部分溶剂,溶液备用。然后采用反相高效液相色谱仪对上述溶液进行分离,色谱条件为:色谱柱为watersnova-pakc18,6μm,190*300mm,流动相a为水:冰醋酸=99.9:0.1,流动相b为乙腈,流速:20ml/min,柱温:20℃,检测波长:244nm,进样量:0.5ml。梯度洗脱如下:时间(min)流动相a比例(%)流动相b比例(%)07525107525304555404555采集相对保留时间为2.3-2.9的制备液,10℃温度下旋干溶剂,得到化合物a5mg,收率0.07%。实施例3:化合物a的检测样品的配制:精密称取依达拉奉原料药10mg,置于50ml容量瓶中,用甲醇:水:冰醋酸=45:55:0.1的混合溶液溶解并定容,配置成依达拉奉含量为0.2mg/ml的样品,4℃控温,半小时内现配现做。色谱条件与检测:仪器:agilent1200hplc色谱柱:agilenteclipsexdb-c18,4.6*150mm(3.5μm);流动相a为甲醇:水:冰醋酸=45:55:0.1,流动相b为甲醇,流速:0.8ml/min,柱温:20℃,检测波长:244nm,进样量:20ml。梯度洗脱如下:时间(min)流动相a比例(%)流动相b比例(%)01000221000523664723664801000检测结果:化合物a出峰时间为17.3min,原料苯肼出峰时间为1.8min。(附图1)实施例4:化合物a的检测样品的配制:精密称取依达拉奉原料药10mg,置于50ml容量瓶中,用乙腈溶解并定容,配置成依达拉奉含量为0.2mg/ml的样品,4℃控温,半小时内现配现做。色谱条件与检测:仪器:agilent1200hplc色谱柱:agilenteclipsexdb-c18,4.6*150mm(3.5μm);流动相a为水:冰醋酸=99.9:0.1,流动相b为乙腈,流速:0.8ml/min,柱温:20℃,检测波长:244nm,进样量:20ml。梯度洗脱如下:时间(min)流动相a比例(%)流动相b比例(%)07525207525605545705545717525检测结果:化合物a出峰时间为27.7min。(附图2)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1