一种2-胺基-3,4-二氢吡喃-3-甲酰胺类似物及其制备方法和用途与流程
本发明提供了涉及药物领域,具体涉及一种2-胺基-3,4-二氢吡喃-3-甲酰胺类似物及其制备方法和用途。
背景技术:
多组分组份反应因为其高收敛性、高原子经济性、高选择性等特性,而被广泛应用到医药小分子化合物的制备中。在绿色化学的大背景下,多组分组份合成不仅提高了整个药化、生化研究效率,更是因为反应步骤的减少,反应试剂消耗的降低而减小了化学合成的环保压力((1).c.hulme,m.ayaz,g.martinez-ariza,f.meddaanda.shaw,insmallmoleculemedicinalchemistry,johnwiley&sons,inc,2015,pp.145-187.(2).d.
3,4-二氢吡喃结构广泛存在于生物活性小分子中,针对含有3,4-二氢吡喃核的化合物的合成一直备受关注。但2-胺基-3,4-二氢吡喃类结构的合成鲜有报导,目前为止,只有两篇文献涉及了其衍生物的制备方法,先后在2011年和2012年发表((8).j.sun,e.-y.xia,q.wuandc.-g.yan,acscomb.sci.2011,13,421-426.(9).b.devibala,s.michaelrajeshands.perumal,greenchem.2012,14,2484-2490.)。文献所提供的方法虽然也是多组分合成,但得到的产物均是2-胺基-3,4-二氢吡喃-3-甲酯,无法一锅法反应完成2-胺基-3,4-二氢吡喃-3-甲酰胺类化合物的制备。2-胺基-3,4-二氢吡喃-3-甲酰胺的合成至今无人报导。因此,对于2-胺基-3,4-二氢吡喃-3-甲酰胺类化合物的制备,如果用文献所报导的方法,具有以下几个缺点:1)文献方法提供的是2-胺基-3,4-二氢吡喃-3-甲酯类衍生物的制备,需要后续酰胺化反应步骤以制备2-胺基-3,4-二氢吡喃-3-甲酰胺化合物,进而增加了制备难度和成本;2)文献所述的方法涉及到了微波反应器的应用,提高了反应成本,限制了制备方法的工业应用;3)文献所述方法用到了炔类化合物作为反应原料,炔类化合物毒性大且各种取代的炔类化合物类别有限,不易获得,一定程度上限制了产物类似物的拓展;4)文献方法反应时间长,而本发明所提供的方法,中间体的生成2小时,而后续反应时间可缩短至5分钟,耗时短;5)文献报导的方法,其产物的提纯有涉及到了柱层析分离,不利于工业化大量生产;6)更重要的是,文献所报导的2-胺基-3,4-二氢吡喃-3-甲酯类化合物均无明确的应用价值。而本发明所制备的2-胺基-3,4-二氢吡喃-3-甲酰胺化合物具有潜在的抗炎活性。
众所周知,适度的炎症反应对机体具有保护作用,但长期或过度的炎症反应会对机体造成损伤,导致疾病发生。一些经典的炎性相关疾病,如(类)风湿性关节炎、动脉粥样硬化、哮喘、炎性肠道疾病、胃炎、肾炎、全身性炎症反应综合征等,其发生和致病过程,均与核转录因子κb(nf-κb)的过度或者持续激活相关。一些外源性因素或者内源性物质通过识别包括toll样受体(toll-likereceptors,tlr)在内的跨膜性蛋白,进入细胞内,经一系列的信号传递,从而激活nf-κb。
toll样受体(tlr)通过识别病原相关分子模式(pamp)或有害的内源性物质诱发天然免疫反应,抵抗病原体感染或组织损伤。tlr家族的tlr2和tlr4在天然免疫和炎症反应中起到重要作用。其中tlr2为家族中识别最多pamps分子的成员,得到广泛关注。tlr2通过识别pamps,致使下游一系列目的基因活化,进而指导目的基因转录和炎性介质的释放。本发明通过定量检测报告基因分泌型胎盘碱性磷酸酶seap(secretedalkalinephosphatase)信号的表达量,来检测tlr2的活化程度,从而反应机体炎症情况。
nf-κb作为各种炎症反应的共同通路,几乎存在于所有细胞中。nf-κb参与了多种细胞因子基因的转录,是细胞核内炎性递质基因转录的开关,在免疫应答、应激反应及细胞凋亡中起着主导作用。通过抑制nf-κb的激活,可对细胞因子的释放进行宏观调控,从而减轻炎症反应造成的脏器损伤。因此,nf-κb成为最具潜力的抗炎药物靶点。((10).a.samuelmawuli,y.michaelbright,a.seth,d.samuel.curr.drugdiscovtechnol,2018,15,1-11.(11).v.pande,,&m.ramos,j.currmedchem,2005,12(3),357-374.(12).h.li,w.han,v.polosukhin,f.e.yull,b.h.segal,c.-mxie,t.s.blackwell.mediatinflamm,2013,503,213.(13).d.vyas,s.samsophear,p.castro,l.chaturvedi,l.azevedo,j.tepe,a.vyas.j.surg.res.,2013,179(2),201.)过度激活的nf-κb形成复合物,从细胞浆内易位进入细胞核诱导趋化因子、粘附分子、免疫识别受体和细胞因子tnf-α,il-6等的转录与表达,最终造成炎症反应。因此通过检测细胞因子的表达量,可以一定程度上检测nf-κb的激活程度,从而判断机体炎症程度。
因此本发明的目的是制备2-胺基-3,4-二氢吡喃-3-甲酰胺类化合物,并提供一种简便高效的合成方法。此外,本发明提供2-胺基-3,4-二氢吡喃-3-甲酰胺化合物在抗炎活性上的数据,以证明本发明所制备的新型化合物的应用价值,更进一步证明本发明所提供的合成方法的价值。
技术实现要素:
本发明一个方面提供了一种制备含有2-胺基-3,4-二氢吡喃-3-甲酰胺结构化合物的制备方法,其包括如下步骤:
1)将双乙烯酮与r2nh2在溶剂中搅拌反应至完全,得到
2)加入r3nh2,r1cho和式a化合物,并加入单质碘反应至反应液完全固化,过滤后将滤饼洗涤干燥得到含有2-胺基-3,4-二氢吡喃-3-甲酰胺结构化合物;
其中式a化合物如下所示,
所得的含有2-胺基-3,4-二氢吡喃-3-甲酰胺结构化合物如式i所示,
x选自c或o;
r1选自氢,c1-c6的烷基,苯基,杂芳基,环烷基,氰基,甲氧基;苯基、杂芳基、环烷基、氰基、甲氧基、萘基、羟基、卤素取代的c1-c6的烷基;c1-c3烷基、苯基、环烷基、氰基、c1-c3烷氧基、卤素、硝基、羟基取代的苯基或萘基;烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的杂芳基;或烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的环烷基;c1-c3取代的芳基、c1-c3取代的杂芳基或c1-c3取代的环烷基;
r2选自氢,c1-c6的烷基,苯基,苄基,杂芳基,环烷基,氰基,甲氧基;苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的c1-c6的烷基;c1-c6烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素、羟基取代的苯基或苄基;烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的杂芳基;或烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的环烷基;c1-c3取代的芳基、c1-c3取代的杂芳基或c1-c3取代的环烷基;
r3选自氢,c1-c6的烷基,苯基,苄基,杂芳基,环烷基,氰基,甲氧基;苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的c1-c6的烷基;c1-c6烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素、羟基取代的芳基或苄基;烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的杂芳基;或烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的环烷基;c1-c3取代的芳基、c1-c3取代的杂芳基或c1-c3取代的环烷基;
r4选自氢,c1-c6的烷基或与r7形成烯键;
r5选自氢,c1-c6的烷基或者和r6形成苯环;
r6选自氢,c1-c6的烷基或者和r5形成苯环;
r7选自氢,c1-c6的烷基或与r4形成烯键。
优选地,本发明中间体在制备过程中不需要分离。
在本发明的技术方案中,制备方法中所用的溶剂选自二氯甲烷,无水乙醇,乙腈。
在本发明的技术方案中,催化剂碘的用量为原料r3nh2用量的5%-30mol%,优选为8-12%。
本发明反应流程如下:
在本发明的技术方案中,所述的式i化合物选自式ii,式iii或式iv所示化合物,
本发明另一个方面提供了一种含有2-胺基-3,4-二氢吡喃-3-甲酰胺结构化合物,其具有如式i所示结构,
x选自c或o;
r1选自氢,c1-c6的烷基,苯基,杂芳基,环烷基,氰基,甲氧基;苯基、杂芳基、环烷基、氰基、甲氧基、萘基、羟基、卤素取代的c1-c6的烷基;c1-c3烷基、苯基、环烷基、氰基、c1-c3烷氧基、卤素、硝基、羟基取代的苯基或萘基;烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的杂芳基;或烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的环烷基;c1-c3取代的芳基、c1-c3取代的杂芳基或c1-c3取代的环烷基;
r2选自氢,c1-c6的烷基,苯基,苄基,杂芳基,环烷基,氰基,甲氧基;苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的c1-c6的烷基;c1-c6烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素、羟基取代的苯基或苄基;烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的杂芳基;或烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的环烷基;c1-c3取代的芳基、c1-c3取代的杂芳基或c1-c3取代的环烷基;
r3选自氢,c1-c6的烷基,苯基,苄基,杂芳基,环烷基,氰基,甲氧基;苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的c1-c6的烷基;c1-c6烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素、羟基取代的芳基或苄基;烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的杂芳基;或烷基、苯基、杂芳基、环烷基、氰基、甲氧基、卤素取代的环烷基;c1-c3取代的芳基、c1-c3取代的杂芳基或c1-c3取代的环烷基;
r4选自氢,c1-c6的烷基或与r7形成烯键;
r5选自氢,c1-c6的烷基或者和r6形成苯环;
r6选自氢,c1-c6的烷基或者和r5形成苯环;
r7选自氢,c1-c6的烷基或与r4形成烯键。
在本发明的技术方案中,所述的式i化合物选自式ii,式iii或式iv所示化合物,
本发明还涉及一些具体的化合物,化合物的结构如下所示:
本发明另一个方面提供了前述式i化合物的单晶;式i化合物为
β=96.495(4)°,
本发明另一各方面提供了前述单晶的制备方法,包括以下步骤:
1)将化合物用乙醇重结晶,过滤后得到的纯品,取纯品化合物,加入四氢呋喃,溶解后过滤,滤液收集;
2)取滤液置于透明样品瓶中,薄膜封口,薄膜上戳小洞静置3-5天后,带有点状颗粒出现,立刻封口,静置1-2周,有块状晶体长成。
本发明另一个方面提供了前述化合物在制备抗炎药物的用途。
附图说明
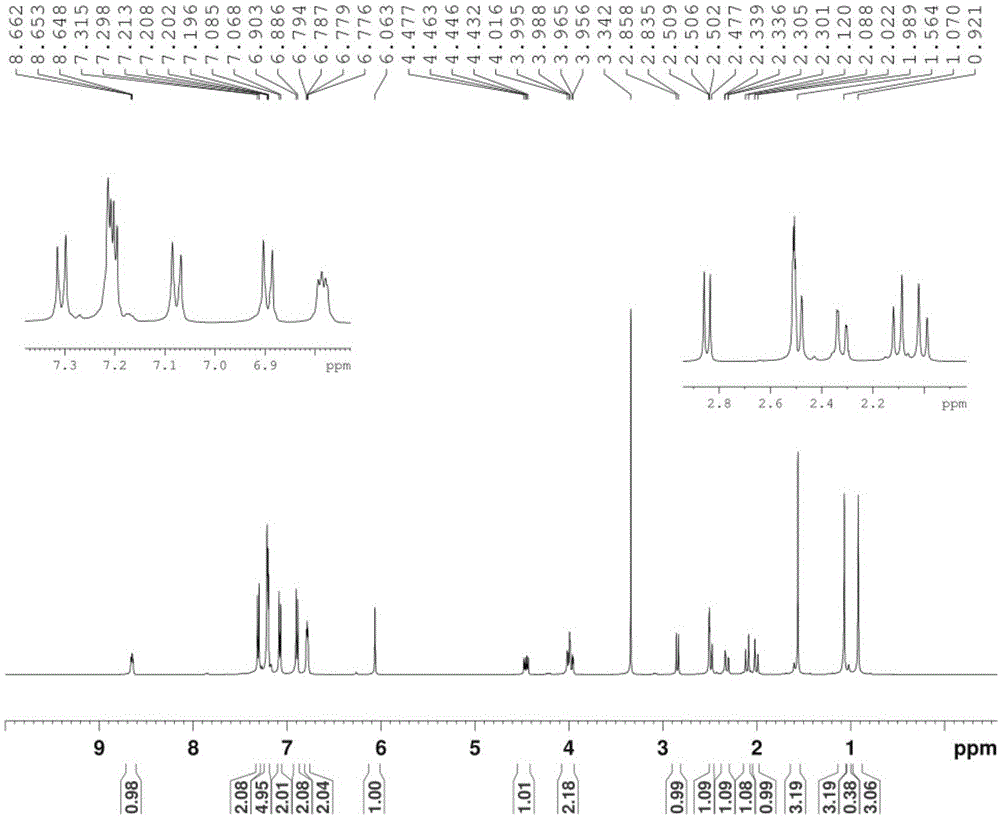
图1为化合物6aaca二维核磁谱图-1hnmr图谱。
图2为化合物6aaca一维核磁谱图-13cnmr图谱。
图3为化合物6aaca二维核磁谱图-hsqc图谱。
图4为化合物6aaca一维核磁谱图-13cdept135图谱。
图5为化合物6aaca二维核磁谱图-cosy图谱。
图6为化合物6aaca二维核磁谱图-hmbc图谱。
图7为代表产物6aaca的xrd单晶结构。
图8为化合物z20(6gcaa)在细胞水平上的抗炎活性评价。
具体实施方式
实施例1制备(2s,3r,4s)-2-(4-甲基苯胺)-n-苄基-4-(4-氯苯基)-3,4,5,6,7,8-六氢-2,7,7-三甲基-5-氧-二氢色烯-3-甲酰胺6aaaa:
在25ml的圆底单口烧瓶中,加入苄胺2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮iv(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和对氯苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,5分钟后,有大量固体生成并迅速固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率95%);熔点166-168℃;1hnmr(500mhz,dmso-d6)δ8.62-8.65(m,nh,1h),7.28-7.30(d,aromatic,2h),7.17-7.19(m,aromatic,3h),7.05-7.08(d,aromatic,2h),6.95-6.98(d,aromatic,2h),6.73-6.76(m,aromatic,4h),5.75(s,nh,1h),4.41-4.48(m,ch,1h),3.92-3.99(m,ch2,2h),2.80-2.84(d,ch,1h),2.29-2.48(m,ch2,2h),2.17(d,ch3,3h),1.95-2.13(m,ch2,2h),1.50(s,ch3,3h),1.06(s,ch3,3h),0.93(s,ch3,3h);13cnmr(125mhz,dmso-d6)δ170.8,167.2,160.1,142.7,142.3,138.8,130.9,130.0,129.8,128.6,127.5,127.3,122.1,117.9,114.9,113.7,88.5,57.0,50.8,42.4,42.0,38.2,31.9,29.3,27.4,23.0,20.6;ms(esi+)m/z543(m+h);anal.calcdforc33h35cln2o3:c,72.98;h,6.50;n,5.16.found:c,72.91;h,6.59;n,5.02.
实施例2制备(2s,3r,4s)-2-(4-氯苯胺)-n-苄基-4-(4-氯苯基)-3,4,5,6,7,8-六氢-2,7,7-三甲基-5-氧-二氢色烯-3-甲酰胺6aaca:
在25ml的圆底单口烧瓶中,加入苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对氯苯胺(2.0mmol)和对氯苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,10分钟后,有大量固体生成,反应混合液逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率71%);熔点168-171℃;1hnmr(500mhz,dmso-d6)δ8.63-8.65(m,nh,1h),7.27-7.31(m,aromatic,2h),7.21(m,aromatic,5h),7.07-7.08(d,aromatic,2h),6.88-6.90(d,aromatic,2h),6.78-6.79(d,aromatic,2h),6.06(s,nh,1h),4.43-4.47(m,ch2,1h),3.96-4.02(m,ch+ch2,2h),2.84-2.86(d,ch,1h),2.30-2.48(m,ch2,2h),1.99-2.12(m,ch2,2h),1.56(s,ch3,3h),1.07(s,ch3,3h),0.92(s,ch3,3h);13cnmr(125mhz,dmso-d6)δ195.5,170.6,166.9,143.7,142.4,138.7,130.9,129.8,129.2,18.5,128.4,127.3,127.1,123.2,118.8,113.7,88.0,56.9,50.7,42.5,41.8,39.5,38.1,32.0,29.0,27.5,26.8,22.8;ms(esi+)m/z563(m+h);anal.calcdforc32h32cl2n2o3:c,68.20;h,5.72;n,4.97.found:c,68.24;h,5.76;n,4.90.
实施例3.(2s,3r,4s)-2-(对甲苯胺)-n-苄基-3,4,5,6,7,8-六氢-2,7,7-三甲基-4-(4-硝基苯基)-5-氧-二氢-色烯-3-甲酰胺6aaae:
在25ml的圆底单口烧瓶中,加入苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和对硝基苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,10分钟后,有大量固体生成,反应混合液逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率81%);熔点180-182℃;1hnmr(300mhz,dmso-d6)δ8.63-8.65(m,nh,1h),8.01-8.11(m,aromatic,2h),7.30-7.31(m,aromatic,2h),6.99-7.14(m,aromatic,5h),6.77(m,aromatic,5h),5.77(s,nh,1h),4.37-4.42(m,ch2,1h),3.92-4.13(m,ch+ch2,2h),2.81-2.87(m,ch,1h),2.19(d,ch3,3h),1.98-2.13(m,2ch2,4h),1.53(s,ch3,3h),1.08(s,ch3,3h),0.96(s,ch3,3h);13cnmr(75mhz,dmso-d6)δ195.1,169.8,151.7,145.7,141.4,138.3,129.4,129.1,128.2,128.1,127.9,127.0,126.8,123.2,117.6,72.3,68.8,56.0,50.1,42.5,41.9,41.6,41.4,41.3,38.3,31.4,28.6,27.0,22.5,20.1;ms(esi+)m/z554(m+h);anal.calcdforc33h35n3o5:c,71.59;h,6.37;n,7.59.found:c,71.63;h,6.31;n,7.48.
实例4.(2s,3r,4s)-2-(对甲苯胺)-n-苄基-4-(4-氰基苯基)-3,4,5,6,7,8-六氢-2,7,7-三甲基-5-氧-二氢-色烯-3-甲酰胺6aaaf:
在25ml的圆底单口烧瓶中,加入苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和对氰基苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,15分钟后,有大量固体生成,反应混合液逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率81%);熔点160-162℃;1hnmr(400mhz,cdcl3)δ7.48(d,j=8.2,aromatic,2h),7.30(d,j=2.0,aromatic,3h),7.25(d,j=8.1,aromatic,2h),7.04(d,j=8.1,aromatic,2h),6.88(t,j=7.4,aromatic,4h),5.74(s,nh,1h),5.50(s,nh,1h),4.45(dd,j=14.6,6.7,ch2,1h),4.22(d,j=11.4,ch,1h),4.11(dd,j=14.6,4.6,ch2,1h),2.48–2.38(m,ch3,3h),2.29(s,ch3,3h),2.14(dd,j=34.9,16.6,ch2,2h),1.64(s,ch3,3h),1.15(s,ch3,3h),1.05(s,ch3,3h).13cnmr(101mhz,cdcl3)δ196.09,170.28,168.10,149.14,141.49,137.01,132.23,129.62,129.34,128.61,128.07,127.74,127.55,118.92,117.82,113.00,109.85,88.56,58.52,50.41,43.45,42.32,39.35,31.56,29.52,27.16,22.76,20.48.hrms:calcdforc34h35n3o3533.2678,found534.2742[m+h].
实例5.(2s,3r,4s)-2-(对甲苯胺)-n-苄基-3,4,5,6,7,8-六氢-4-(2,4-二氯苯基)-2,7,7-三甲基-5-氧-二氢-色烯-3-甲酰胺6aaah:
在25ml的圆底单口烧瓶中,加入苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和2,4-二氯苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,24小时后,有大量固体生成,反应混合液逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率47%);熔点160.3-161.9℃;1hnmr(400mhz,cdcl3)δ=7.33(d,j=2.1,aromatic,1h),7.25(s,aromatic,1h),7.24(d,j=3.6,aromatic,1h),7.04(d,j=8.1,aromatic,4h),6.94(d,j=8.4,aromatic,1h),6.88(d,j=8.1,aromatic,4h),6.09–5.88(m,2nh,2h),4.75(d,j=11.5,ch,1h),4.52–4.45(m,ch2,1h),4.05(dd,j=14.8,4.4,ch2,1h),2.54(d,j=11.5,ch,1h),2.28(d,j=12.5,ch3+ch2,5h),1.96(d,j=21.0,ch2,2h),1.62(s,ch3,3h),1.08(s,ch3,3h);13cnmr(101mhz,cdcl3)δ=196.50,195.94,170.79,170.08,167.67,167.36,141.78,141.72,139.87,137.06,136.95,136.09,135.17,132.97,132.17,131.49,130.20,129.54,129.42,129.22,129.03,128.60,127.59,127.54,127.46,127.35,127.27,127.22,118.06,117.70,114.15,112.29,88.99,88.19,77.31,77.00,76.68,57.98,52.53,50.47,50.31,43.62,43.35,42.41,42.28,39.91,34.39,31.53,31.40,29.58,29.26,27.84,27.01,22.99,22.43,20.49;lc-ms/ms(esi+)m/z599.243(m+na).
实施例6.(2s,3r,4s)-2-(对甲苯胺)-n-苄基-3,4,5,6,7,8-六氢-4-(4-羟基苯基)-2,7,7-三甲基-5-氧-二氢-色烯-3-甲酰胺6aaai:
在25ml的圆底单口烧瓶中,加入苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和对羟基苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,50分钟后,有大量固体生成,反应混合液逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率72%);熔点162-164℃;1hnmr(400mhz,dmso)δ9.15(s,oh,1h),8.69–8.51(m,nh,1h),7.20(d,j=6.9,aromatic,3h),6.98(d,j=8.0,aromatic,2h),6.88(d,j=8.1,aromatic,2h),6.80(d,j=6.6,aromatic,2h),6.74(d,j=8.0,aromatic,2h),6.65(d,j=8.2,aromatic,2h),5.74(s,nh,1h),4.44(dd,j=15.4,7.0,ch2,1h),4.04–3.95(m,ch,1h),3.89(d,j=11.0,ch2,1h),2.84(d,j=11.2,ch,1h),2.48–2.25(m,ch2,2h),2.19(s,ch3,3h),2.05(dd,j=35.8,9.8,ch2,2h),1.51(s,ch3,3h),1.08(s,ch3,3h),0.95(s,ch3,3h).13cnmr(101mhz,dmso)δ195.38,171.19,166.12,155.94,155.43,142.32,138.75,133.44,129.85,128.80,128.47,128.23,127.29,127.07,117.62,115.25,114.58,88.35,57.52,50.79,42.27,41.89,37.78,31.70,29.26,27.20,22.95,20.53.anal.calcdforc33h36n2o4:c,75.55;h,6.92;n,5.34.found:c,75.59;h,6.84;n,5.41.
实施例7.(2s,3r,4s)-2-(对甲苯胺)-n-苄基-3,4,5,6,7,8-六氢-2,7,7-三甲基-4-(2-萘基)-5-氧-二氢-色烯-3-甲酰胺6aaaj:
在25ml的圆底单口烧瓶中,加入苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和乙萘醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,5分钟后,有大量固体生成,反应混合液逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率68%);熔点157-159℃;1hnmr(400mhz,cdcl3)δ7.82–7.71(m,aromatic,4h),7.46(dd,j=5.9,3.3,aromatic,2h),7.27-7.31(m,aromatic,1h),7.05(d,j=7.0,aromatic,3h),6.95–6.81(m,aromatic,4h),6.51(d,j=7.6,aromatic,2h),5.91(s,nh,1h),5.41(s,nh,1h),4.34(dd,j=14.5,7.8,ch2+ch,2h),4.09–3.98(m,ch2,1h),2.63(d,j=11.4,ch,1h),2.50(d,j=19.2,ch2,2h),2.30(s,ch3,3h),2.09(dd,j=33.8,16.2,ch2,2h),1.19(s,ch3,3h),1.05(d,j=8.6,ch3,3h).13cnmr(101mhz,cdcl3)δ195.93,170.67,166.86,142.88,141.37,136.66,132.17,128.90,128.80,128.68,128.50,127.74,127.49,124.44,118.39,113.73,87.91,59.26,50.56,43.62,42.21,38.71,31.60,29.37,27.22,22.59.hrms:calcdforc37h38n2o3558.2882,found559.2945[m+h].
实施例8.(2s,3r,4s)-n-(2-氟苯基)-2-(对甲苯胺)-4-(4-氯苯基)-3,4,5,6,7,8-六氢-2,7,7-三甲基-5-氧-二氢-色烯-3-甲酰胺6daaa:
在25ml的圆底单口烧瓶中,加入2-氟苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料2-氟苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和对氯苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,120分钟后,有大量固体生成,反应混合液逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率76%);熔点166-168℃;1hnmr(500mhz,dmso-d6)δ8.65(m,nh,1h),7.24-7.29(m,aromatic,3h),6.98-7.12(m,aromatic,6h),6.74-6.76(d,aromatic,2h),6.60-6.63(m,aromatic,1h),5.72(s,nh,1h),4.36-4.40(m,ch2,1h),3.95-4.11(m,ch+ch2,2h),2.82-2.84(d,ch,1h),2.31-2.47(m,ch2,2h),2.19(s,ch3,3h),1.99-2.16(m,ch2,2h),1.51(s,ch3,3h),1.07(s,ch3,3h),0.95(s,ch3,3h);13cnmr(126mhz,dmso)δ195.51,170.95,167.19,161.29,159.35,142.52,130.81,130.18,129.91,129.84,129.66,129.47,129.40,128.44,125.49,125.38,124.40,121.50,117.89,115.50,115.33,113.62,88.49,56.96,50.72,41.99,38.16,36.25,36.22,31.87,29.18,28.41,27.38,22.91,20.59;ms(esi+)m/z561(m+h);anal.calcdforc33h34clfn2o3:c,70.64;h,6.11;n,4.99.found:c,70.72;h,6.10;n,4.95.
实施例.(2s,3r,4s)-2-(对甲苯胺)-4-(4-氯苯基)-3,4,5,6,7,8-六氢-2,7,7-三甲基-5-氧-n-对甲苯基-二氢-色烯-3-甲酰胺6eaaa:
在25ml的圆底单口烧瓶中,加入对甲苯胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和对氯苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,8小时后,有大量固体生成,反应混合液逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体。白色固体(产率83%);熔点185-187℃;1hnmr(500mhz,dmso-d6)δ9.95(m,nh,1h),7.26-7.47(m,aromatic,4h),6.98-7.19(m,aromatic,6h),6.78-6.79(d,aromatic,2h),5.73(s,oh,1h),4.05-4.07(m,ch,1h),2.91-2.93(d,ch,1h),2.28-2.36(m,ch2,2h),2.19(s,ch3,3h),2.15(s,ch3,3h),1.99-2.12(m,ch2,2h),1.58(s,ch3,3h),1.04(s,ch3,3h),0.96(s,ch3,3h);13cnmr(126mhz,dmso)δ195.30,169.64,167.54,142.72,142.18,135.42,133.15,129.90,129.58,128.50,127.23,123.54,123.37,120.59,118.01,113.56,88.29,57.83,50.69,42.02,38.30,31.94,29.11,27.51,23.01,20.90,20.60.ms(esi+)m/z543(m+h);anal.calcdforc33h35cln2o3:c,72.98;h,6.50;n,5.16.found:c,72.93;h,6.58;n,6.63.
实施例10制备(2s,3r,4s)-(对甲苯胺)-n-丁基-4-(4-氯苯基)-3,4,5,6,7,8-六氢-2,7,7-三甲基-5-氧-二氢色烯-3-甲酰胺6faaa:
在25ml的圆底单口烧瓶中,加入正丁胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料正丁胺反应完全,中间体生成后,加入双甲酮(2.0mmol),对甲基苯胺(2.0mmol)和对氯苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,20分钟后,有大量固体生成,逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体,白色固体用无水乙醇重结晶。白色固体(产率71%);熔点146-148℃;1hnmr(500mhz,dmso-d6)δ8.05(m,nh,1h),7.27-7.29(d,aromatic,2h),6.97-7.04(m,aromatic,4h),6.74-6.75(d,aromatic,2h),5.83(s,nh,1h),3.91-3.93(d,ch,1h),3.07-3.08(d,ch,1h),2.68-2.83(m,ch2,2h),2.31-2.47(m,ch2,2h),2.19(s,ch3,3h),1.99-2.13(m,ch2,2h),1.49(s,ch3,3h),1.11-1.16(m,ch2,2h),1.08(s,ch3,3h),0.88-0.95(m,ch2+ch3,5h),0.73-0.76(m,ch3,3h);13cnmr(125mhz,dmso-d6)δ195.5,170.6,167.3,142.7,142.4,30.7,130.4,19.9,129.5,128.3,117.8,113.6,88.5,57.0,50.7,42.0,38.6,38.2,31.9,31.2,29.2,27.4,22.9,20.6,19.6,14.0;ms(esi+)m/z509(m+h);anal.calcdforc30h37cln2o3:c,70.78;h,7.33;n,5.50.found:c,70.71;h,7.39;n,5.57.
实施例11制备(2s,3r,4s)-2-(对甲苯胺)-n-苄基-3,4,5,6,7,8-六氢-4-(4-甲氧基苯基)-2-甲基-5-氧-二氢色烯-3-甲酰胺6abad:
在25ml的圆底单口烧瓶中,加入苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入1,3-环己二酮(2.0mmol),对甲基苯胺(2.0mmol)和对甲氧基苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,3分钟后,有大量固体生成,逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体,白色固体用无水乙醇重结晶。白色固体(产率41%);熔点161.9-163.1℃;1hnmr(400mhz,cdcl3)δ7.27–7.20(m,aromatic,3h),7.11(d,j=8.3,aromatic,2h),7.03(d,j=8.0,aromatic,2h),6.88(d,j=8.2,aromatic,4h),6.79(d,j=8.3,aromatic,2h),5.84(s,nh,1h),5.44(d,j=5.2,nh,1h),4.41(dd,j=14.7,6.3,ch2,1h),4.20–4.10(m,ch+ch2,2h),3.78(d,j=7.2,ch3,3h),2.64–2.40(m,ch3,3h),2.33–2.15(m,ch3+ch2,5h),1.97(dd,j=12.4,9.1,ch2,2h),1.63(s,ch3,3h);13cnmr(100mhz,cdcl3)δ=196.13,171.19,168.30,158.09,141.91,137.05,134.91,129.48,128.84,128.51,128.07,127.58,117.72,115.73,113.99,88.23,59.83,55.09,43.54,38.36,36.84,28.70,22.77,20.44,20.02;hrms:calcdforc32h34n2o4510.2519,found511.2578[m+h].
实施例12制备(2s,3r,4s)-4-(4-氯苯)-2-甲基-5-氧-n-(对甲苯基)-2-(对甲苯胺)-2,3,4,5-四氢吡喃[3,2-c]色烯-3-甲酰胺,6ecaa:
在5ml的反应瓶中,加入对甲苯胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料对甲苯胺反应完全,中间体生成后,加入4-羟基香豆素(2.0mmol),对甲基苯胺(2.0mmol)和对氯苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,15分钟后,有大量固体生成,逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体,白色固体用无水乙醇重结晶。白色固体(产率85%);熔点172.9-176.8℃;1hnmr(400mhz,cdcl3)δ8.01(d,j=7.7hz,aromatic,1h),7.50(t,j=7.7hz,aromatic,1h),7.36(t,j=7.4hz,aromatic,1h),7.17(d,j=7.9hz,aromatic,3h),7.10(d,j=12.9hz,aromatic,6h),6.97(d,j=7.4hz,aromatic,3h),6.86(d,j=7.7hz,aromatic,2h),5.92(s,nh,1h),4.48(d,j=11.4hz,ch,1h),2.82(d,j=11.4hz,ch,1h),2.31(s,ch3,3h),2.21(s,ch3,3h),1.83(s,ch3,3h);13cnmr(101mhz,cdcl3)δ168.88,162.47,160.76,153.00,140.86,139.81,135.34,133.59,132.77,131.81,129.96,129.56,129.47,128.96,128.49,128.46,124.04,122.64,121.07,118.68,116.73,115.68,104.45,90.07,59.43,53.52,39.75,22.76,20.84,20.43;hrms:calcdforc34h29cln2o4564.1816,found565.1876[m+h].
实施例13制备(2s,3r,4s)-4-(4-氯苯)-2-甲基-5-氧-n-(苯基)-2-(对甲苯胺)-2,3,4,5-四氢吡喃[3,2-c]色烯-3-甲酰胺,6gcaa:
在5ml的反应瓶中,加入苯胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料对甲苯胺反应完全,中间体生成后,加入4-羟基香豆素(2.0mmol),对甲基苯胺(2.0mmol)和对氯苯甲醛ix(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,1分钟后,有大量固体生成,逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体,白色固体用无水乙醇重结晶。白色固体(产率91%);熔点191.8-193.6℃;1hnmr(400mhz,dmso)δ10.19(s,nh,1h),8.08(d,j=7.7,aromatic,1h),7.67(t,j=7.5,aromatic,1h),7.48(d,j=7.6,aromatic,1h),7.42(t,j=8.9,aromatic,3h),7.31(d,j=8.3,aromatic,4h),7.24(d,j=8.1,aromatic,2h),7.10(t,j=7.2,aromatic,1h),7.02–6.81(m,aromatic,4h),6.02(s,nh,1h),4.50(d,j=11.4,ch,1h),3.24(d,j=11.4,ch,1h),2.14(s,ch3,3h),1.83(s,ch3,3h);13cnmr(101mhz,dmso)δ168.88,160.18,158.08,152.79,141.16,140.83,138.04,132.64,131.47,129.79,129.69,129.25,129.00,128.70,124.85,123.20,120.47,118.44,116.81,115.64,104.65,90.28,57.57,38.93,22.89,20.48;hrms:calcdforc33h27cln2o4550.1659,found549.1587[m-h].
实施例14制备(2s,3r,4s)-n-苯基-4-(4-氯苯基)-2,7-二甲基-5-氧-2-(对甲苯胺)-2,3,4,5-四氢吡喃[4,3-b]吡喃-3-甲酰胺,6adaa:
在5ml的反应瓶中,加入苄胺(2.0mmol),低温下搅拌,缓慢滴入化合物双乙烯酮(2.0mmol),搅拌中回温到室温,继续搅拌2小时,tlc检测跟踪,待原料苄胺反应完全,中间体生成后,加入4-羟基-6-甲基吡喃酮(2.0mmol),对甲基苯胺(2.0mmol)和对氯苯甲醛(2.0mmol),以及单质碘(0.2mmol),室温下持续搅拌,30分钟后,有大量固体生成,逐渐固化,终止反应,加入2ml的乙腈,减压抽滤,滤饼依次用饱和硫代硫酸纳溶液和水洗涤,然后干燥,得到白色固体,白色固体用无水乙醇重结晶。白色固体(产率78%);熔点180.5-182.1℃;1hnmr(400mhz,cdcl3)δ7.24(d,j=1.7hz,aromatic,3h),7.17(d,j=8.4hz,aromatic,2h),7.07(d,j=8.3hz,aromatic,2h),7.01(d,j=8.1hz,aromatic,2h),6.83(d,j=8.2hz,aromatic,4h),5.90(s,aromatic,1h),5.82(s,2nh,2h),4.43(dd,j=14.8,6.7hz,ch2,1h),4.27(d,j=11.5hz,ch,1h),4.08(dd,j=14.8,4.5hz,ch2,1h),2.53(d,j=11.5hz,ch2,1h),2.25(s,ch3,3h),2.14(s,ch3,3h),1.62(s,ch3,3h);13cnmr(101mhz,cdcl3)δ170.26,162.89,162.48,161.63,141.28,139.79,136.78,132.56,129.57,128.85,128.64,127.64,127.44,118.02,101.41,100.26,89.12,58.85,43.57,38.93,22.65,20.47,19.80;hrms:calcdforc31h29cln2o4528.1816,found529.1883[m+h].
含有2-胺基-3,4-二氢吡喃-3-甲酰胺结构的新型化合物i,ii和iii的抗炎活性评价(活性数据见实例15和实例16)
实施例15tlr2介导的seap信号抑制活性评价:
a.quanti-blu碱性磷酸酶活性检测:
hek-bluehtlr2细胞首先在加有10%胎牛血清和1%青霉素-链霉素双抗的dmem培养基中培养。随后接种到96孔板中(每孔200μl培养基,接种量为4x104细胞)在37℃恒温过夜。第二天换液,然后依次加入100μl药物溶液,50μl含有10ng/ml终浓度的pam3csk4,在37℃恒温培养24h,并设立空白和阳性对照。第三天取50μl培养基上清至新的96孔板中并加入50μlquanti-blue(invivogen)缓冲液。静置15分钟,最后在620nm波长下下测定吸光值。
b.mtt细胞活性检测:
第一天接种hek-bluehtlr2细胞如96孔板(40,000/孔,200μl含10%胎牛血清和1%青霉素-链霉素双抗的dmem培养基),37℃,5%co2培养24小时。第二天换液并加入待测化合物和10ng/ml终浓度的pam3csk4,在37℃,5%co2培养24小时。第三天每孔加入15μlmtt溶液(pbs溶解),在37℃培养3小时,随后弃去培养基并干燥过夜。最后每孔加入100μldmso并摇晃混匀30分钟,在570nm波长下测定吸光值。
经过活性筛选,100μm的6ecaa和6gcaa对seap信号的抑制率达到90%以上(表1),虽然用量偏大,但该剂量下并没有明显的细胞毒性。将化合物6ecaa和6gcaa分别命名为z19和z20,进一步测试得到其ic50,其结果见表1。因此,可以明确化合物z19和z20具有潜在的抗炎活性。
表1.靶向nf-κb的tlr2介导的seap信号抑制活性结果[a]
a10ng/mlpam3csk4作为阳性对照.b三次独立操作后的平均值.
为进一步明确其抗炎活性,我们在此基础上,对化合物z19和z20在细胞水平上评价其对nf-κb激活后导致的炎症因子表达量的影响。初步评价结果显示,z19和z20均能够有效抑制激活后的炎症因子tnf-α和il-6的表达,而同时z20的效果更好。因此单独针对z20,扩大样本量,评价其对被激活的炎症因子tnf-α和il-6的表达水平的影响,具体见实例16。
实施例16脂多糖lps(lipopolysaccharide)激活nf-κb后导致炎症因子tnf-α和白介-6(il-6)的表达抑制率检测
两组成熟raw264.7细胞株同时给予lps刺激,lps组采用含有dmso的dmem培养细胞,lps+z20组用含有新型抗炎药物的dmem培养细胞。lps和吡喃衍生物z20的工作浓度分别为100ng/ml、100μmol/l。两组刺激维持6h后,弃细胞培养上清,用2mldpbs清洗细胞两遍,每孔加入500μltrion裂解液反复吹打收集细胞,提取细胞rna。
q-pcr检测raw264.7细胞中炎症因子mrna的表达:分别提取lps组和lps+z20组的rna,经逆转录转为cdna,而后经q-pcr扩增,根据ct值对两组细胞进行炎症因子mrna水平检测。
在lps刺激raw264.7巨噬细胞系后,用阴性对照物dmso(二甲亚枫)和z20共处理被lps刺激后的细胞,6小时后,通过实时荧光定量pcr实验检测两组炎症因子的mrna表达水平。
结果显示lps+新型抗炎药z20组相比于lps组,tnf-α的mrna表达水平可以降低0.59倍,il-6的表达水平可以降低0.43倍(图8)。由此我们得到初步结论:z20能够有效抑制lps导致的炎症因子释放,明确表现出抗炎效果。
实施例17单晶培养
对代表化合物6aaca进行单晶培养:将产物用乙醇重结晶,得到的纯品,取纯品化合物6aaca10mg,加入四氢呋喃(thf),溶解后过滤,滤液收集。取3ml滤液置于5ml的透明样品瓶中,薄膜封口,毛细管在薄膜上戳3-5个小洞。静置3-5天后,带有点状颗粒出现,立刻封口(旋紧样品瓶盖子),静置1-2周,有块状晶体长成。凡士林包裹取出的晶体待测。将晶体进行xrd单晶衍射检测,解析单晶数据,确定产物结构(单晶数据见下面数据)xrd单晶结构见图7.
化合物6aaca的晶体数据和结构精修.
- 还没有人留言评论。精彩留言会获得点赞!
- 2,4-O-二-α-L-吡喃鼠李糖基-β-D-吡喃葡萄糖基薯蓣皂甙的合成方法
- 一类6-芳基-3-环氨基甲基吡喃酮类衍生物的制备及用途的制作方法
- 吡喃葡糖氧基吡唑衍生物及其在药物中的应用的制作方法
- 取代亚甲基吡喃酮类衍生物及其制备方法和用途的制作方法
- 6-芳基-3-取代亚甲基吡喃酮类化合物及其制备方法和用途的制作方法
- 吡喃葡糖氧基吡唑衍生物、含该衍生物的药物组合物及其制备中的中间体的制作方法
- 7-糖氧基苯并吡喃衍生物及含有该衍生物作为活性成分的抗变应性剂的制作方法
- 取代的氨甲基苯并二氢吡喃在运动障碍和治疗锥体束外运动障碍药物引发的不利影响的 ...的制作方法
- 氨甲基取代的2,3-二氢吡喃并[2,3-b]吡啶类,制备方法和在医药中的应用的制作方法
- 3-甲基-八氢环庚三烯并吡喃-2-酮及其制备方法