一种基于ddRAD方法的新品种鉴定方法与流程
本发明属于生物技术领域,具体涉及一种基于ddrad方法的新品种鉴定方法。
背景技术:
近些年来,许多品种的育种亲本越来越集中在少数优良品种或品系上,使所选育的新品种在植物学性状上很相似,同种异名、同名异种现象日益严重,难以区分,而且随着品种贸易的快速发展,对植物新品种权益进行保护的重要性也随之越来越突出,如:在审定新品种以及授予新品种的品种权前要进行品种鉴定,确定该品种的特异性、一致性和稳定性;在发生品种权纠纷案件时需要对争议品种进行鉴定,判断品种权的归属;在引进外来品种时必须快速鉴定和精确的描述该新品种的性状特征,确保引种的成功性;对种苗有必要实施早期鉴定。另外,在现阶段中国市场条件下,种苗市场管理不完善,以劣充优、以假乱真现象屡有发生,使花费几年到几十年的时间,投入大量技术、劳力、物质资源及资金培育出新品种的育种者、育种单位和公司以及种植伪劣苗木的农民蒙受难以弥补的经济损失,严重阻碍了农业生产的发展。因此,如何有效地鉴定植物品种成为一个非常重要的问题卜,而要实现有效地鉴定植物品种就需要一种具有高度准确性的植物品种鉴定方法。
近年蓬勃发展的高通量测序可以作为基因型分析、品种鉴定的一种新策略。目前研究者已经提出了若干基于二代测序技术的简化代表文库测序策略,其中最简单的方法是分离纯化特定片段大小范围的限制性酶切片段。rad(restrictionsite-associateddna,限制性酶切位点关联dna)测序方法,该技术利用限制性内切酶对物种基因组进行酶切,从而回收一定大小的片段来构建基因文库,然后进行测序。其具有高通量、低成本、实验周期较短且无需参考基因组等优势,因此得到广泛的应用。且由于rad标记的是在全基因组序列中呈现特异性酶切位点附近的短dna片段,反映了整个基因组的特征,因此通过rad-seq能够在绝大多数的生物中获得数以万计的基因组水平上由单个核苷酸的变异所引起的dna序列多态性位点,简称为单核苷酸多态性位点(snps)。而ddrad(double-digestrad,双酶切rad)测序是rad衍生的一种测序策略,所谓双酶切是通过将一种识别位点较少的限制性内切酶和另一种识别位点较多的常见酶相结合,对基因组dna进行酶切,这样处理能够免去物理打断的过程,可直接进行目的片段的筛选,并通过pcr扩增在第二个酶切位点附近引入个体识别标签(index),从而使得更多的样本能够混合在一起进行高通量测序。这个方法能够在获得更多的有效数据的同时大大减少实验成本,且由于双酶切对dna文库的筛选更为严格,序列两端必须为不同的酶切且其长度在某一区间,如500bp左右的片段,因此测出的序列也更加准确。ddrad测序与rad的单末端测序相比,ddrad的双末端测序能够增加标记的特异性和准确性。近年,在花生、草莓及油菜等物种中已经利用该测序方法构建了若干高密度遗传图谱。由于rad-seq技术具有操作简便,周期短,实验成本低,不受参考基因组的限制,能够覆盖到全基因组范围上,一次实验即获得的大量snp信息,因而,可以用于任何物种的高密度图谱的构建、定位高分化区域及功能基因、筛选分子标记、品种鉴定及群体遗传分析等优点。
技术实现要素:
本发明的目的在于提供一种基于ddrad方法的新品种鉴定方法,该方法提供了多产、稳定的待测品种基因组,以及低成本、高效、高质量的ddrad测序方法,能够实现基因检测多样性,有效提高鉴别结果的准确性和稳定性。
本发明为实现上述目的所采取的技术方案为:
一种基于ddrad方法的新品种鉴定方法,包括基因组提取和ddrad方法测序及分析鉴定,其中ddrad测序方法中双酶切反应用含有巯基乙胺和2-吡啶甲醇的体系进行酶切处理。本发明通过对待测品种的基因组提取后,采用ddrad方法降低和简化其基因组的复杂度,并对特定的酶切片段进行高通量测序后,与可供参考的基因组文库进行分析比对,以此鉴定区别出新品种,能够有效提高鉴别结果的准确性和稳定性。
作为优选,双酶切体系中加入巯基乙胺和2-吡啶甲醇后,能快速打开dna的识别序列,将处于对称轴内部的切割位点暴露出来,使得酶活性中心更容易接触到识别位点,从而提高酶切速率,同时两者能与dna提取物中残留的乙醇、氯仿形成之间共价连接,降低残留物对内切酶的影响,从而协助内切酶进行特异性识别,确定目标序列切割点的均一性,进而降低了异常位点的产生,减小了等位基因缺失对基因检测多样性的影响,增加了酶切以及后续分析鉴定的准确性和稳定性。
作为优选,ddrad方法测序包括如下步骤:
1)双酶切反应:利用两种常见性限制性内切酶对dna基因组进行酶切,得到限制性酶切片段;
2)连接反应:使用t4dna连接酶对酶切所得限制性片段连接上dna条形码接头a1和a2,得到形如“a1接头-dna插入片段-a2接头”的序列对;
3)核心片段回收选择:将连接产物进行琼脂糖凝胶电泳分离回收得到目标dna片段;
4)pcr扩增:对回收的含有不同条形码的目标dna片段进行pcr扩增并富集;
5)磁珠纯化:对pcr扩增产物采用磁珠法纯化;
6)片段分布检查:对纯化dna目的片段采用低熔点琼脂糖凝胶电泳进行片段分布检查和质量检测;
7)测序:将纯化dna目的片段稀释后进行高通量测序,获得简化后待测品种基因组信息。
进一步优选,双酶切反应中两种限制性内切酶为4~6碱基常见限制性内切酶,且不含稀有碱基限制性内切酶,即限制性内切酶kpnⅰ和ecorⅰ。限制性内切酶kpnⅰ的专一识别顺序是5’-ggtac↓c-3’,ecorⅰ的专一识别顺序是5’-g↓aattc-3’,“↓”表示限制性内切酶的酶切位点。
再进一步优选,双酶切反应的具体步骤如下:取0.5ml离心管,配制50μlkpnⅰ和ecorⅰ双酶切体系,加入1μlkpnⅰ、5μl10×lbuffer、0.2~0.3μl巯基乙胺、0.2~0.3μl2-吡啶甲醇和1μgdna提取物,其余体积用超纯水补齐,置于35~39℃恒温水浴1~1.5h后,加入1μlecorⅰ、5μlbsa、和5μl10×hbuffer,置于35~39℃恒温水浴1~1.5h后,在60~65℃下失活15~20min,4~5℃保存酶切产物,用1%琼脂糖凝胶电泳检测酶切结果。bsa是一种牛血清白蛋白,可以通过提高溶液中蛋白质的浓度,防止酶的分解和非特异性吸附,减轻某些酶的变性,从而对酶起保护作用,广泛应用于限制性内切酶酶切反应实验中。
进一步优选,连接反应中a1接头由正链和负链组成:
正链:5’tacggcgaccaccgagatcacacxxxxx3’,
负链:5’gwcyyyyygtgtgatctcggtggtcgccgta3’,
其中,正链中的xxxxx代表dna条形码序列,负链中的yyyyy代表与dna条形码互补序列,w代表碱基a或t;上述a1接头中dna条形码序列由12个4~9碱基序列组成,分别为:
attgc、ggtga、tggct、tagcc、gagtcg、ctacag、cctgtag、attcgca、gctacgct、tgctaagt、ctgaagctg、gtggcctag。
进一步优选,连接反应中a2接头由正链和负链组成:
正链:5’cgatagagcctcttagc3’,
负链:5’gtgactgtgtgcccgatctat3’。
再进一步优选,连接反应具体步骤为:取40μl酶切产物,加入1~1.5μla1接头和3.5~4.5μla2接头,0.5μlt4dna连接酶和4.0~5.0μl10×t4连接酶缓冲液,在23~25℃连接反应1.5~2h,60~65℃失活15~20min,4~5℃保存连接产物。
进一步优选,核心片段回收选择的步骤如下:用2.0%琼脂糖凝胶电泳分离,电泳缓冲液为1xtae,电泳电压为4v/cm,电泳时间为85~95min,切割得到300~500bp片段。
进一步优选,pcr扩增所用引物碱基序列为:
p1:agcgttccaccgtagaga,
p2:ccaatccggcnnnnnataacaac,
其中,引物序列p2中的nnnnn代表index序列,上述index序列包括:gccaat、atcacg。
进一步优选,pcr反应程序为:98℃预变性3min,进入循环,98℃变性10s,56℃退火30s,72℃延伸30s,共18个循环,然后72℃延伸15min,10℃降温10min,最终保持4℃恒温。
其中,pcr扩增体系配制如下:分别取20~22μl回收的dna片段,1.0~1.5μl引物p1,1.0~1.5μl引物p2,0.6~1.0μl10mmdntp,0.6~1.0μlq5hfdna聚合酶,22~24μlq5反应缓冲液混合,再加入1~6μldh2o定容总反应体积为50μl。
进一步优选,磁珠纯化的具体步骤为:取45μlpcr扩增产物,加入5μl分子级别的水和50μl充分混匀后的磁性微珠,置于室温下10~15min;再移至到磁性架上,待磁性微珠聚集吸附到管壁一侧后,将上清液全部移除,然后加入80~100μl100%乙醇,清洗2~3次后,移除所有的乙醇,自然风干;再加入15μl分子级别的水,将富集后的目的片段洗脱下来,即得纯化dna目的片段,备用。
进一步优选,片段分布检查的条件设定如下:将纯化dna目的片段用2%的低熔点琼脂糖凝胶电泳进行质量检测,跑胶电压为120v,时间为85~95min,测定片段范围。
进一步优选,测序时将纯化dna目的片段先稀释至10nm,再进行上机测序。ddrad方法中采用两种限制性内切酶处理基因组dna,酶切标签数目提升,起始dna用量较低,并且采用更为精准的核酸电泳和片段回收系统,进行片段大小的选择,可以降低片段选择过程对相同位点在文库间代表性的影响,使得片段大小均匀性提高,进而个体间分型重复性和分型准确性均有提高,达到了数据可利用率和准确性高,性价比高的目的。
作为优选,分析鉴定是将待测品种的测序结果性状相似品种进行基因分析,并得出物种树,即可鉴定出新品种。本发明中采用ddrad方法可以降低和简化待测品种基因组的复杂度,并对特定的酶切片段进行高通量测序后,与可供参考的基因组文库进行遗传与变异分析及比对,且能够不受基因组序列的限制,对没有参考基因组的物种也可以进行大规模筛查,以此鉴定区别出新品种,有效提高鉴别结果的准确性和稳定性。
作为优选,基因组提取步骤为:将待测品种细胞进行裂解处理后,再利用溶剂进行抽提,除杂后得到待测品种dna基因组。在裂解植物细胞的基础上,多次利用有机溶剂进行抽提,可以使蛋白质等沉淀于有机试剂中,而核酸保留在水相,从而达到分离核酸的目的,提取dna的流程简便,易于操作,且dna提取得率高,稳定性优异。
为优化上述方案,基因组提取具体采取的措施如下:
1)预处理:称取15~25mg植物待测品种干材料,加入2~5mg石英砂研磨成细粉末状,将粉末转移到2.0ml离心管中;
2)细胞裂解:加入0.5~0.9ml缓冲液混匀后60~65℃水浴90~120min,水浴过程中颠倒混匀4~5次,颠倒混匀时操作需轻缓,然后在10000~15000xg的条件下离心10~12min,吸取上清液并置于新的2.0ml离心管中,即得裂解液,上述缓冲液配制100ml配方如下:10ml1.0mol/ltris-hcl、5ml0.5mol/ledta-na2、28ml5.0mol/lnacl、3gctab、2gpvp-40t、100μlβ-巯基乙醇、1g抗环血酸,余量加dh2o定容;
3)提取:向裂解液中加入0.6~0.9ml氯仿异戊醇溶液(氯仿:异戊醇=24:1,v/v),颠倒混匀8~12min,然后在10000~12000xg的条件下离心10~12min,吸取上清液并置于新的1.5ml离心管中,重复操作至离心后两个液面之间没有沉淀,即得粗提液;
4)沉淀:向粗提液中加入0.3~0.6ml预冷异丙醇,轻轻混匀,-15~-22℃下放置18~22min,然后10000~12000xg离心8~10min,弃上清液,再离心2~4min收集剩余液体,用移液器吸除剩余液体,即得粗提物;
5)除杂:向粗提物中加入0.1~0.2ml100mg/lrnase,35~38℃下放置30~60min,然后加入0.1~0.2ml去离子水、0.1~0.2ml5mol/lnacl溶液、0.8~1.0ml预冷95%乙醇,轻轻混匀,然后10000~12000xg离心8~10min,弃上清液,即得精提物;
6)冲洗:向精提物中加入0.5~0.7ml75%乙醇,弹起沉淀,然后10000~12000xg离心2~3min,弃上清液,重复1~2次后,风干乙醇,即得到dna提取物。
本发明的有益效果为:
1)本发明中采用细胞裂解后再利用溶剂多次提取的方法快速提取dna基因组,其方法简便,易于操作,目的性和实用性强,且dna提取速率和得率高,稳定性优异;
2)本发明中采用ddrad方法可以大幅度地提高双酶切的速率,降低待测品种基因组的复杂情况,利用较低的成本就可以获得足够准确和稳定的结果,为研究其基因组的序列差异提供有利工具,能实现精确基因分型,开展遗传解析;
3)本发明中基于ddrad方法进行新品种的鉴定,有效解决了因品种间遗传差异逐渐缩小而造成的鉴定难度增大的问题,且能提供给遗传与变异解析的信息量也很大,尤其适用于鉴别遗传差异相对较小,鉴别难度较大的植物样品。
本发明采用了上述技术方案提供一种基于ddrad方法的新品种鉴定方法,弥补了现有技术的不足,设计合理,操作方便。
附图说明
图1为实施例4的物种树示意图。
具体实施方式
以下结合实施例和附图对本发明作进一步详细描述:
实施例1:
一种基于ddrad方法的新品种鉴定方法,包括基因组提取和ddrad方法测序及分析鉴定,其中ddrad测序方法中双酶切反应用含有巯基乙胺和2-吡啶甲醇的体系进行酶切处理,该方法通过对待测品种的dna基因组提取后,采用ddrad方法降低和简化其基因组的复杂度,并对特定的酶切片段进行高通量测序后,与可供参考的基因组文库进行分析比对,以此鉴定区别出新品种,能够有效提高鉴别结果的准确性和稳定性。
双酶切体系中加入巯基乙胺和2-吡啶甲醇后,能快速打开dna的识别序列,将处于对称轴内部的切割位点暴露出来,使得酶活性中心更容易接触到识别位点,从而提高酶切速率,同时两者能与dna提取物中残留的乙醇、氯仿形成之间共价连接,降低残留物对内切酶的影响,从而协助内切酶进行特异性识别,确定目标序列切割点的均一性,进而降低了异常位点的产生,减小了等位基因缺失对基因检测多样性的影响,增加了酶切以及后续分析鉴定的准确性和稳定性。
dna基因组基于ddrad方法测序,具体包括如下步骤:
1)双酶切反应:利用两种常见性限制性内切酶对dna基因组进行酶切,得到限制性酶切片段;
2)连接反应:使用t4dna连接酶对酶切所得限制性片段连接上dna条形码接头a1和a2,得到形如“a1接头-dna插入片段-a2接头”的序列对;
3)核心片段回收选择:将连接产物进行琼脂糖凝胶电泳分离回收得到目标dna片段;
4)pcr扩增:对回收的含有不同条形码的目标dna片段进行pcr扩增并富集;
5)磁珠纯化:对pcr扩增产物采用磁珠法纯化;
6)片段分布检查:对纯化dna目的片段采用低熔点琼脂糖凝胶电泳进行片段分布检查和质量检测;
7)测序:将纯化dna目的片段稀释后进行高通量测序,获得简化后待测品种基因组信息。
双酶切反应中两种限制性内切酶为4~6碱基常见限制性内切酶,且不含稀有碱基限制性内切酶,即限制性内切酶kpnⅰ和ecorⅰ。限制性内切酶kpnⅰ的专一识别顺序是5’-ggtac↓c-3’,ecorⅰ的专一识别顺序是5’-g↓aattc-3’,“↓”表示限制性内切酶的酶切位点。
双酶切反应的具体步骤如下:取0.5ml离心管,配制50μlkpnⅰ和ecorⅰ双酶切体系,加入1μlkpnⅰ、5μl10×lbuffer、0.2μl巯基乙胺、0.2μl2-吡啶甲醇和1μgdna提取物,其余体积用超纯水补齐,置于35℃恒温水浴1h后,加入1μlecorⅰ、5μlbsa、和5μl10×hbuffer,置于35℃恒温水浴1h后,在60℃下失活15min,4℃保存酶切产物,用1%琼脂糖凝胶电泳检测酶切结果。bsa是一种牛血清白蛋白,可以通过提高溶液中蛋白质的浓度,防止酶的分解和非特异性吸附,减轻某些酶的变性,从而对酶起保护作用,广泛应用于限制性内切酶酶切反应实验中。
连接反应中a1接头由正链和负链组成:
正链:5’tacggcgaccaccgagatcacacxxxxx3’,
负链:5’gwcyyyyygtgtgatctcggtggtcgccgta3’,
其中,正链中的xxxxx代表dna条形码序列,负链中的yyyyy代表与dna条形码互补序列,w代表碱基a或t;上述a1接头中dna条形码序列由12个4~9碱基序列组成,分别为:
attgc、ggtga、tggct、tagcc、gagtcg、ctacag、cctgtag、attcgca、gctacgct、tgctaagt、ctgaagctg、gtggcctag。
连接反应中a2接头由正链和负链组成:
正链:5’cgatagagcctcttagc3’,
负链:5’gtgactgtgtgcccgatctat3’。
连接反应具体步骤为:取40μl酶切产物,加入1μla1接头和3.5μla2接头,0.5μlt4dna连接酶和4.0μl10×t4连接酶缓冲液,在23℃连接反应1.5h,60℃失活15min,4℃保存连接产物。
核心片段回收选择的步骤如下:用2.0%琼脂糖凝胶电泳分离,电泳缓冲液为1xtae,电泳电压为4v/cm,电泳时间为85min,切割得到300~500bp片段。
pcr扩增所用引物碱基序列为:
p1:agcgttccaccgtagaga,
p2:ccaatccggcnnnnnataacaac,
其中,引物序列p2中的nnnnn代表index序列,上述index序列包括:gccaat、atcacg。
pcr反应程序为:98℃预变性3min,进入循环,98℃变性10s,56℃退火30s,72℃延伸30s,共18个循环,然后72℃延伸15min,10℃降温10min,最终保持4℃恒温。
其中,pcr扩增体系配制如下:分别取20μl回收的dna片段,1.0μl引物p1,1.0μl引物p2,0.6μl10mmdntp,0.6μlq5hfdna聚合酶,22μlq5反应缓冲液混合,再加入4.8μldh2o定容总反应体积为50μl。
磁珠纯化的具体步骤为:取45μlpcr扩增产物,加入5μl分子级别的水和50μl充分混匀后的磁性微珠,置于室温下10min;再移至到磁性架上,待磁性微珠聚集吸附到管壁一侧后,将上清液全部移除,然后加入80μl100%乙醇,清洗2次后,移除所有的乙醇,自然风干;再加入15μl分子级别的水,将富集后的目的片段洗脱下来,即得纯化dna目的片段,备用。
片段分布检查的条件设定如下:将纯化dna目的片段用2%的低熔点琼脂糖凝胶电泳进行质量检测,跑胶电压为120v,时间为85min,测定片段范围。
测序时将纯化dna目的片段先稀释至10nm,再进行上机测序。ddrad方法中采用两种限制性内切酶处理基因组dna,酶切标签数目提升,起始dna用量较低,并且采用更为精准的核酸电泳和片段回收系统,进行片段大小的选择,可以降低片段选择过程对相同位点在文库间代表性的影响,使得片段大小均匀性提高,进而个体间分型重复性和分型准确性均有提高,达到了数据可利用率和准确性高,性价比高的目的。
分析鉴定是将待测品种的测序结果与性状相似品种进行基因分析,并得出物种树,即可鉴定出新品种。本发明中采用ddrad方法可以降低和简化待测品种基因组的复杂度,并对特定的酶切片段进行高通量测序后,与可供参考的基因组文库进行遗传与变异分析及比对,且能够不受基因组序列的限制,对没有参考基因组的物种也可以进行大规模筛查,以此鉴定区别出新品种,有效提高鉴别结果的准确性和稳定性。
基因组提取步骤为:将待测品种细胞进行裂解处理后,再利用溶剂进行抽提,除杂后得到待测品种dna基因组。在裂解植物细胞的基础上,多次利用有机溶剂进行抽提,可以使蛋白质等沉淀于有机试剂中,而核酸保留在水相,从而达到分离核酸的目的,提取dna的流程简便,易于操作,且dna提取得率高,稳定性优异。
其中,提取dna基因组具体采取的措施如下:
1)预处理:称取15mg植物待测品种干材料,加入2mg石英砂研磨成细粉末状,将粉末转移到2.0ml离心管中;
2)细胞裂解:加入0.5ml缓冲液混匀后60℃水浴90min,水浴过程中颠倒混匀4次,颠倒混匀时操作需轻缓,然后在10000xg的条件下离心10min,吸取上清液并置于新的2.0ml离心管中,即得裂解液,上述缓冲液配制100ml配方如下:10ml1.0mol/ltris-hcl、5ml0.5mol/ledta-na2、28ml5.0mol/lnacl、3gctab、2gpvp-40t、100μlβ-巯基乙醇、1g抗环血酸,余量加dh2o定容;
3)提取:向裂解液中加入0.6ml氯仿异戊醇溶液(氯仿:异戊醇=24:1,v/v),颠倒混匀8min,然后在10000xg的条件下离心10min,吸取上清液并置于新的1.5ml离心管中,重复操作至离心后两个液面之间没有沉淀,即得粗提液;
4)沉淀:向粗提液中加入0.3ml预冷异丙醇,轻轻混匀,-15℃下放置18min,然后10000xg离心8min,弃上清液,再离心2min收集剩余液体,用移液器吸除剩余液体,即得粗提物;
5)除杂:向粗提物中加入0.1ml100mg/lrnase,35℃下放置30min,然后加入0.1ml去离子水、0.1ml5mol/lnacl溶液、0.8ml预冷95%乙醇,轻轻混匀,然后10000xg离心8min,弃上清液,即得精提物;
6)冲洗:向精提物中加入0.5ml75%乙醇,弹起沉淀,然后10000xg离心2min,弃上清液,重复1次后,风干乙醇,即得到dna提取物。
实施例2:
一种基于ddrad方法的新品种鉴定方法,包括基因组提取和ddrad方法测序及分析鉴定。
基因组提取具体采取的措施如下:
1)预处理:称取20mg植物待测品种干材料,加入3mg石英砂研磨成细粉末状,将粉末转移到2.0ml离心管中;
2)细胞裂解:加入0.7ml缓冲液混匀后65℃水浴100min,水浴过程中颠倒混匀5次,颠倒混匀时操作需轻缓,然后在15000xg的条件下离心10min,吸取上清液并置于新的2.0ml离心管中,即得裂解液,上述缓冲液配制100ml配方如下:10ml1.0mol/ltris-hcl、5ml0.5mol/ledta-na2、28ml5.0mol/lnacl、3gctab、2gpvp-40t、100μlβ-巯基乙醇、1g抗环血酸,余量加dh2o定容;
3)提取:向裂解液中加入0.8ml氯仿异戊醇溶液(氯仿:异戊醇=24:1,v/v),颠倒混匀10min,然后在12000xg的条件下离心10min,吸取上清液并置于新的1.5ml离心管中,重复操作至离心后两个液面之间没有沉淀,即得粗提液;
4)沉淀:向粗提液中加入0.5ml预冷异丙醇,轻轻混匀,-20℃下放置20min,然后12000xg离心10min,弃上清液,再离心4min收集剩余液体,用移液器吸除剩余液体,即得粗提物;
5)除杂:向粗提物中加入0.1ml100mg/lrnase,37℃下放置50min,然后加入0.2ml去离子水、0.2ml5mol/lnacl溶液、1.0ml预冷95%乙醇,轻轻混匀,然后12000xg离心10min,弃上清液,即得精提物;
6)冲洗:向精提物中加入0.7ml75%乙醇,弹起沉淀,然后12000xg离心3min,弃上清液,重复2次后,风干乙醇,即得到dna提取物。
dna基因组基于ddrad方法进行测序,包括如下步骤:
1)双酶切反应:利用两种常见性限制性内切酶对dna基因组进行酶切,得到限制性酶切片段,即限制性内切酶kpnⅰ和ecorⅰ,限制性内切酶kpnⅰ的专一识别顺序是5’-ggtac↓c-3’,ecorⅰ的专一识别顺序是5’-g↓aattc-3’,“↓”表示限制性内切酶的酶切位点,双酶切反应的具体步骤如下:取0.5ml离心管,配制50μlkpnⅰ和ecorⅰ双酶切体系,加入1μlkpnⅰ、5μl10×lbuffer、0.3μl巯基乙胺、0.3μl2-吡啶甲醇和1μgdna提取物,其余体积用超纯水补齐,置于37℃恒温水浴1.5h后,加入1μlecorⅰ、5μlbsa、和5μl10×hbuffer,置于37℃恒温水浴1h后,在65℃下失活20min,4℃保存酶切产物,用1%琼脂糖凝胶电泳检测酶切结果;
2)连接反应:使用t4dna连接酶对酶切所得限制性片段连接上dna条形码接头a1和a2,得到形如“a1接头-dna插入片段-a2接头”的序列对,其具体步骤为:取40μl酶切产物,加入1μla1接头和3.5μla2接头,0.5μlt4dna连接酶和4.0μl10×t4连接酶缓冲液,在25℃连接反应2h,65℃失活15min,4℃保存连接产物,其中,a1接头由正链和负链组成:
正链:5’tacggcgaccaccgagatcacacxxxxx3’,
负链:5’gwcyyyyygtgtgatctcggtggtcgccgta3’,
其中,正链中的xxxxx代表dna条形码序列,负链中的yyyyy代表与dna条形码互补序列,w代表碱基a或t,上述a1接头中dna条形码序列由12个4~9碱基序列组成,分别为:
attgc、ggtga、tggct、tagcc、gagtcg、ctacag、cctgtag、attcgca、gctacgct、tgctaagt、ctgaagctg、gtggcctag,
连接反应中a2接头由正链和负链组成:
正链:5’cgatagagcctcttagc3’,
负链:5’gtgactgtgtgcccgatctat3’;
3)核心片段回收选择:将连接产物进行琼脂糖凝胶电泳分离回收得到目标dna片段,其步骤如下:用2.0%琼脂糖凝胶电泳分离,电泳缓冲液为1xtae,电泳电压为4v/cm,电泳时间为90min,切割得到300~500bp片段;
4)pcr扩增:对回收的含有不同条形码的目标dna片段进行pcr扩增并富集,pcr扩增所用引物碱基序列为:
p1:agcgttccaccgtagaga,
p2:ccaatccggcnnnnnataacaac,
其中,引物序列p2中的nnnnn代表index序列,上述index序列包括:index1为gccaat,index2为atcacg,
pcr反应程序为:98℃预变性3min,进入循环,98℃变性10s,56℃退火30s,72℃延伸30s,共18个循环,然后72℃延伸15min,10℃降温10min,最终保持4℃恒温,其中,pcr扩增体系配制如下:分别取22μl回收的dna片段,1.0μl引物p1,1.0μl引物p2,0.8μl10mmdntp,0.8μlq5hfdna聚合酶,23μlq5反应缓冲液混合,再加入1.4μldh2o定容总反应体积为50μl;
5)磁珠纯化:对pcr扩增产物采用磁珠法纯化,其具体步骤为:取45μlpcr扩增产物,加入5μl分子级别的水和50μl充分混匀后的磁性微珠,置于室温下15min;再移至到磁性架上,待磁性微珠聚集吸附到管壁一侧后,将上清液全部移除,然后加入100μl100%乙醇,清洗3次后,移除所有的乙醇,自然风干;再加入15μl分子级别的水,将富集后的目的片段洗脱下来,即得纯化dna目的片段,备用;
6)片段分布检查:对纯化dna目的片段采用低熔点琼脂糖凝胶电泳进行片段分布检查和质量检测,其条件设定如下:将纯化dna目的片段用2%的低熔点琼脂糖凝胶电泳进行质量检测,跑胶电压为120v,时间为90min,测定片段范围;
7)测序:将纯化dna目的片段稀释至10nm后进行高通量测序,获得简化后待测品种基因组信息。
对待测品种的dna基因组基于ddrad方法进行分析鉴定是将待测品种的测序结果与性状相似品种进行基因分析,并得出物种树,即可鉴定出新品种。
实施例3:
对待测品种的dna基因组基于ddrad方法进行测序进一步优化措施如下:对dna基因组提取的细胞裂解步骤进行优化,具体步骤如下:加入0.7ml缓冲液混匀后65℃水浴100min,水浴过程中颠倒混匀5次,颠倒混匀时操作需轻缓,然后在15000xg的条件下离心10min,吸取上清液并置于新的2.0ml离心管中,即得裂解液,上述缓冲液配制100ml配方如下:10ml1.0mol/ltris-hcl、5ml0.5mol/ledta-na2、28ml5.0mol/lnacl、3gctab、2gpvp-40t、100μlβ-巯基乙醇、1g抗环血酸、0.5g牛磺酸、0.6g茶碱,余量加dh2o定容,植物细胞中含有多糖、多酚等次生物质含量较高,次生物质包裹着dna,使dna提取较为困难,牛磺酸和茶碱在水浴时彼此形成搭接,能对缓冲液裂解细胞产生增益作用,能促使多糖和多酚的官能团与茶碱嘌呤环上7位n上的h发生替换,从而使多糖、多酚解开对dna的包裹,释放出更多的dna,增加dna的提取速率和得率,同时将核纤层蛋白的中间纤维打破,并对dna的碱基产生保护,使dna的完整性不被破坏,避免产生开环及断裂,增加dna提取物的稳定性。
其他步骤和方法与实施例2相同,以此来进行新品种的鉴定。
实施例4:
选取一种新型变异形态的非洲菊作为待测品种,将待测样品分为2组,分为优化组和常规组,其中常规组对非洲菊dna提取物进行双酶切反应时,其所用双酶切体系中未含有巯基乙胺和2-吡啶甲醇,每组设2个平行试验,每个平行取2份样品进行测定,测序平台为illuminahiseq2500,将数据进行统计分析后得到结果。
利用实施例2中方法对非洲菊样品进行鉴定,检测及鉴定结果如下:优化组样品共产生185.78m条原始reads,其中,质量得分≥20的碱基占97.56%,质量得分≥30的碱基占89.12%,说明所得reads为高质量reads,即指带有正确barcode和酶切位点序列,以及未出现adaptor序列、ns和质量小于10的碱基;统计后发现,含有接头的reads的比例较低,平均为2.13%,含有正确酶切位点的比例平均为98.57%,整体都保持了高质量的酶切标准;而常规组样品共产生173.24m条原始reads,其中,质量得分≥20的碱基占95.67%,质量得分≥30的碱基占81.52%,说明所得reads为较高质量reads,即指带有正确barcode和酶切位点序列,以及未出现adaptor序列、ns和质量小于10的碱基;统计后发现,含有接头的reads的比例较优化组高,平均为6.75%,含有正确酶切位点的比例平均为93.72%,可见,优化组所得的测序结果较常规组更具有准确性和稳定性。
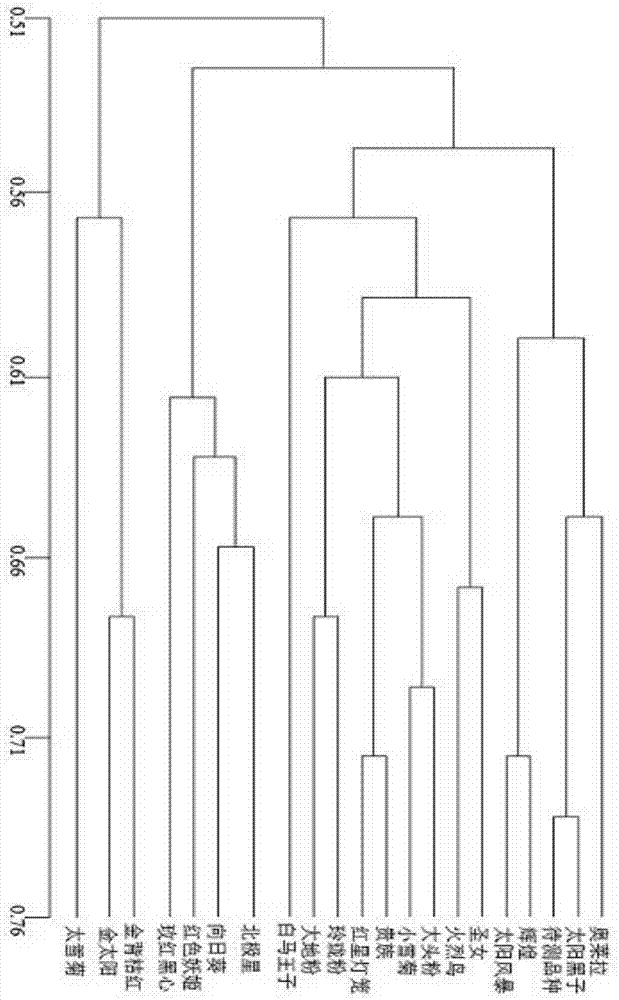
将优化组的测序结果与性状相似的20个品种进行基因分析,并得出物种树,见说明书附图1,从附图中可得,待测品种为非洲菊新品种与已知品种太阳黑子为近似品种,两者的相似系数大于0.74。
上述实施例中的常规技术为本领域技术人员所知晓的现有技术,故在此不再详细赘述。
以上实施方式仅用于说明本发明,而并非对本发明的限制,本领域的普通技术人员,在不脱离本发明的精神和范围的情况下,还可以做出各种变化和变型。因此,所有等同的技术方案也属于本发明的范畴,本发明的专利保护范围应由权利要求限定。
- 还没有人留言评论。精彩留言会获得点赞!