一种一次分离多个已知序列DNA片段的未知侧翼序列的方法与流程
本发明属于基因工程技术领域,具体涉及一种一次分离多个已知序列dna片段的未知侧翼序列的方法
背景技术:
分离一段已知序列dna片段的未知侧翼序列是基因组学和分子生物学研究中常用的实验,比如,在全基因组序列未知的情况下分离一个基因上游的启动子序列;分离bac/yac/fosmid文库中插入的外源片段的两个末端序列;分离农杆菌介导的转基因物种中t-dna在基因组上的插入位点以及植物中内源转座子重新转座后在基因组上的插入位点(例如分离水稻的tos17或玉米的mu、ac/ds转座子突变体的侧翼序列);在医学研究中,分离基因治疗时所用载体在基因组上的插入位点。围绕这些实验需求,目前已报道了多种分离侧翼序列的方法,常用的方法有tail-pcr(thermalasymmetricinterlacedpcr)及与其相关的一系列改进技术(liuetal.1995,liuandchen2007,wangetal.2011)、反向pcr(inversepcr)、接头连接pcr(adapterligation-mediatedpcr)(o'malleyetal.2007,leonietal.2011),pcrwalking(zhangetal.2014)等。这些方法的本质都是在已知序列dna片段的旁侧引入一个可以与位于已知序列上的特异引物组合、能够进行pcr扩增的元件,从而可以扩增已知序列dna片段旁侧的未知序列,例如,tail-pcr中使用的一个24-26bp的随机简并引物使之有可能在已知序列dna片段的旁侧某一位置退火并和位于已知序列上的特异引物组合进行pcr扩增;反向pcr是将包含已知序列dna片段和旁侧待分离的片段环化后,利用其已知序列自身的正向和反向引物配对扩增已知序列dna片段的侧翼序列(ochmanetal.1988);而接头连接pcr则是将基因组dna随机打断,再利用dna连接酶在已知序列dna片段的附近连接上一个合成的dna接头序列,使其可以和位于已知序列dna片段上的特异引物组合并扩增待分离的dna片段(krawczyketal.2011)。
上述tail-pcr、反向pcr和接头连接pcr等方法存在着下述共同缺陷:第一,由于技术设计方面的固有缺陷,实验的成功率不高。例如,tail-pcr是否成功不仅取决于三轮pcr的效率,更取决于已知序列dna片段上游大约500bp的范围内是否有所用随机简并引物的结合位点,有时,需要换用不同的随机简并引物才能扩增出目标片段。另外,接头连接pcr和反向pcr都需要连接酶进行dna之间的连接或环化,连接反应的效率一直是限制此类方法效率的关键原因;第二,这些方法实验流程繁琐,实验周期长。例如,接头连接pcr需要进行dna的酶切、纯化、环化后才能进行pcr扩增;第三,一次反应只能分离一个已知序列dna片段一侧(t-dna或tos17元件的左侧或右侧)的侧翼序列,应用于大量样品时费时费力;第四,这些方法的最终pcr产物都需要利用第一代测序技术sanger测序来完成,成本高、效率低。在新一代测序技术应用日益广泛的今天,亟需开发一种基于新一代测序技术的高通量方法,用于分离已知序列dna片段的侧翼序列。
技术实现要素:
本发明的目的是利用新一代高通量测序手段高效率的分离已知序列dna片段的侧翼序列,并且可以从左、右两端同时分离同一个样品中多个已知序列dna片段的侧翼序列。
一次分离多个已知序列dna片段的未知侧翼序列的方法,包括如下步骤:
(1)利用tn5转座酶复合体将基因组dna随机打断,同时,利用tn5在切割dna的同时能够连接上一段dna接头序列的特性,在dna断点处加上gps-seq接头;
(2)反应完成后纯化dna,pcr反应之前的热变性过程会使全部的dna都变性为单链dna;
(3)变性后,所有的dna片段根据是否含有已知序列dna片段而分为两部分:不含已知序列dna片段元件的部分和含有已知序列dna片段元件的部分;在第一个pcr循环时,只有和已知序列dna片段互补配对的特异性引物sp_l1可以和模板链退火并线性扩增,而不含已知序列dna片段由于没有引物结合位点,不会被扩增;
(4)在第二个pcrcycle时,以第一个cycle扩增的产物为模板,以sp_l1与n7xx为引物,进行第二个cycle的扩增;随后,继续以sp_l1与n7xx为引物进行随后的pcr扩增,直到第一轮pcr结束;
(5)进行第二轮pcr扩增,以第一轮pcr产物为模板,以带illuminap5接头序列的嵌套引物sp_l2和ppmi7引物进行pcr扩增;
(6)进行第三轮pcr扩增,以第二轮pcr产物为模板,利用引物ppmi7和n5xx,进行pcr扩增。
所述gps-seq接头由两条退火在一起的dna单链组成,它们作为tn5转座酶的反应底物在反应后被连接到基因组dna上,并作为后续pcr反应的引物结合位点。
所述引物n7xx的5’端为ppmi7序列,中间为8bp的index序列,3’端碱基序列可退火结合在gps-seq接头序列上。
所述嵌套引物sp_l2的3’端碱基序列可以在sp_l1的左侧退火。
所述引物n5xx的3’端碱基结合在sp_l2的illuminap5接头序列上,中间为8bp的index序列,5’端碱基序列为ppmi5。
经过三轮pcr扩增,已知序列dna片段的侧翼序列得到极大富集,同时,illumina测序平台所需的接头序列和index序列也全部加在了pcr产物上,进而满足后续的ta克隆测序或高通量测序需求。
本发明巧妙利用了tn5转座酶复合体可以在打断dna的同时,将dna接头连接到打断的dna片段上的特性,理论上,基因组dna只要能够被打断,其断点处都会被连接上dna接头,连接dna接头的效率近乎100%,远远高于反向pcr、接头pcr等同类技术。结合第二代高通量测序技术,可以同时分离一个样品中多个已知序列dna片段的侧翼序列,而且可以从其左右两侧同时进行。对同一个已知序列dna片段,同时从左右两端分离其侧翼序列,可以提高侧翼序列的分离效率,如果能够同时分离到其左右两侧的侧翼序列,它们可以互相验证,提高准确度。
本发明的有益效果:(1)操作简单。gps-seq利用转座酶tn5复合体通过一步反应在打断dna的同时,在其两端断点处上加上dna接头,避免了目前常用的tail-pcr、反向pcr和接头pcr等操作中繁琐的步骤,是一种简单的侧翼序列分离方法。(2)通量灵活。gps-seq在应用上的通量可高可低,可以用于一份样品中一段已知序列dna片段侧翼序列的分离,也可以用于一份样品中多个已知序列dna片段侧翼序列的分离;结合二代高通量测序的barcoding技术,不同样品中的pcr产物可以混合在一起进行测序,是一种高效的侧翼序列分离方法。(3)结果可靠。结合二代高通量测序,对于同一个侧翼序列,gps-seq可以同时得到多条测序产物,提高了侧翼序列的准确性。此外,从同一个已知序列dna片段的两端同时分离侧翼序列,不仅可以提高成功率,而且两端的结果还可以互相验证,提高可靠性,是一种准确的侧翼序列分离方法。
附图说明
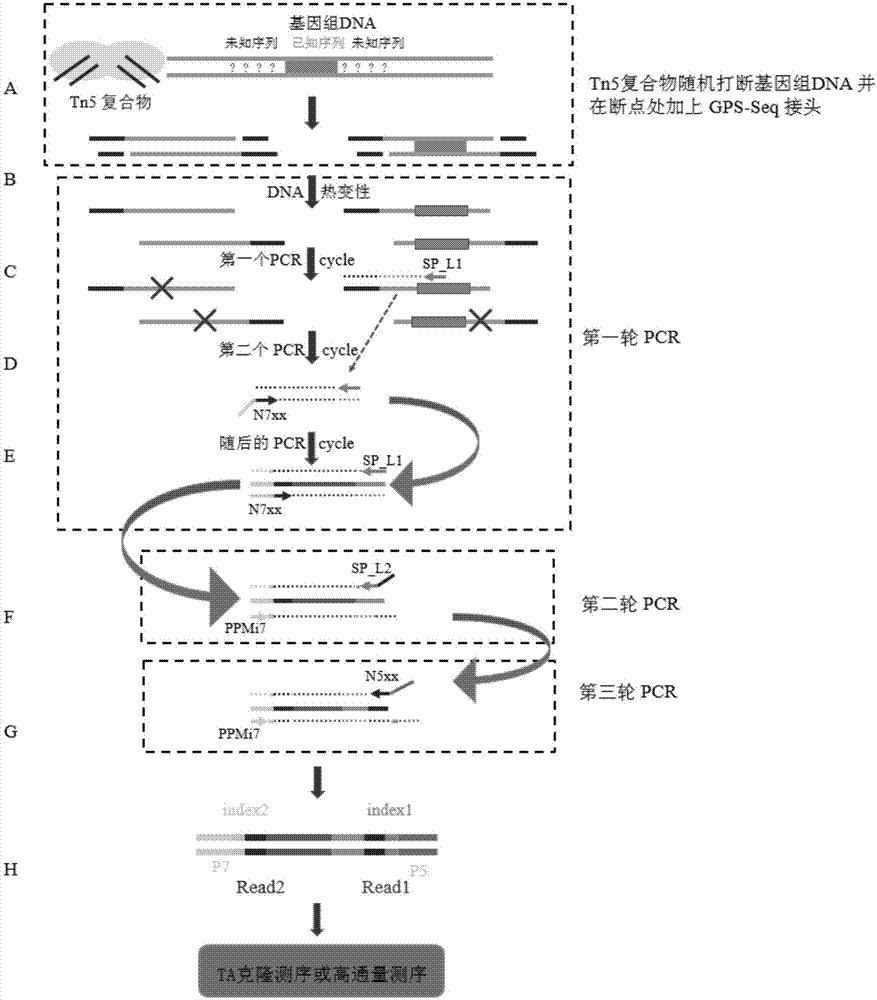
图1为gps-seq分离侧翼序列的主要流程(注:虚线表示pcr扩增时新合成的链,带箭头的实线表示引物,×表示该dna片段不能被扩增)。
图2为gapdh片段及其侧翼序列扩增的引物结合位置图(注:引物sp_gapdhrv1(结合在gapdh基因1680-1704bp处)和sp_gapdhrv2(结合在gapdh基因1679-1655bp处)分别作为第一次和第二次pcr扩增时退火在gapdh基因已知序列片段上的锚定引物;引物n702为第一轮pcr时所用的引物,其包含三部分,5’端为引物ppmi7,中间部分为8bp的index序列,3’端可以退火结合在gps-seq接头上。引物ppmi7是第二次pcr扩增时所用引物,只有第一次pcr扩增时被n702特异扩增的pcr产物才能被ppmi7扩增。引物fw(结合在gapdh基因1259-1283bp处)是利用qpcr检验gapdh基因1655-1704bp片段的上游区域是否被富集时与引物sp_gapdhrv2配对的引物)。
图3为gapdh基因目的片段的q-pcr结果,从中可以看出,目标区段被极大的富集。
图4为tos17侧翼序列插入t载体后的ta克隆电泳检测图;注:图中marker(m)条带分别100bp,200bp,300bp,400bp,500bp,600bp,700bp,800bp,900bp,1000bp及1500bp,其中最亮的条带为500bp。编号表示第二轮pcr的产物连入t载体再转化大肠杆菌后长出的单克隆;41为空白对照。
图5为gps-seq的数据分析流程图。
图6为突变体k664的t-dna插入位置验证电泳图(此处及以下电泳图marker均为tiangenmarkeriii,条带分别为200bp、500bp、800bp、1200bp、2000bp、3000bp、4500bp)。
图7为突变体k664的tos17插入位置验证电泳图(一)。
图8为突变体k664的tos17插入位置验证电泳图(二)。
图9为k664的包含sp_fl_cdnal2与sp_fl_cdnar2引物的reads分别与水稻日本晴参考mrna序列及重组载体的比对情况。
图10为突变体k664超表达的fl_cdna序列扩增电泳图。
具体实施方式
下面结合具体实施例对本发明做进一步说明。
下述本发明的三个实例所用的tn5转座酶复合体的制备方法根据truepreptmadvanceddnasampleprepkit,50ngdna(南京诺唯赞生物科技有限公司,catno.s301-05/01-b)说明书中描述的方法操作。所用接头序列为primera:5'-phos-ctgtctcttatacacatct-nh3-3'与primerc:5′-gtctcgtgggctcggagatgtgtataagagacag-3′,接头的名字命名为gps-seqdna接头。
本发明方法的原理如图1所示。
实施例1利用q-pcr验证水稻gapdh片段的侧翼序列可以被gps-seq富集
为了验证设计的gps-seq方法是否可行,以水稻基因组中的gapdh基因的1655-1704bp片段为例,尝试分离其上游的dna区域,并利用q-pcr检验该片段的上游区域是否被富集。步骤如下:
1.1转座酶复合体打断水稻基因组并在dna片段断点处连接gps-seq接头
利用tn5复合体打断水稻基因组dna,按表1成分在灭菌pcr管中配置反应体系,然后将反应管置于pcr仪中,运行如下反应程序:105℃热盖,55℃10min,10℃hold。
表1.转座酶复合体反应体系
1.2利用磁珠法对转座酶复合体打断后的产物进行纯化
所用的磁珠为vahtstmdnacleanbeads(vazyme,n411-01/02/03),根据打断后的dna片段大小,采用1.8×beads纯化,即50μl打断产物中加入90μl(1.8×50)磁珠,洗脱体积为30μl,取2μl用
1.3利用gps-seq特异性扩增gapdh片段的侧翼序列
实验中用到的gapdh基因,扩增gapdh片段的侧翼序列的引物结合位置如图2所示。
pcr扩增所需引物序列及引物结合位置信息如下:
表2.pcr扩增gapdh片段所需引物序列及引物结合位置信息
注:下划线表示样品特定的index序列。
按表3、表4成分在灭菌的pcr管中配置两轮pcr反应体系(反应所用的酶货号为:kapa,kk2102)并在pcr仪中运行表5设置的反应程序,扩增gapdh片段的侧翼序列。
表3.第一轮pcr反应体系
注:template1为水稻基因组dna经转座酶打断、加接头,纯化后的产物。
表4.第二轮pcr反应体系
注:template2为第一轮pcr的反应产物,取2μl稀释50倍后的稀释液。
表5.第一轮与第二轮pcr的反应程序
1.4纯化pcr扩增后的产物
反应完成后,然后将两次pcr的反应终产物,同样用vazyme公司vahtstmdnacleanbeads分别纯化,选用1.8×beads纯化,纯化步骤及浓度测定方法同1.2。
1.5q-pcr检测pcr产物中gapdh基因上游片段的富集程度
为了检测gps-seq方法对gapdh片段的侧翼序列的富集程度,我们取两轮pcr纯化后的产物,以水稻actin基因(正向引物f(5’→3’):cttcataggaatggaagctgcgggta;反向引物r(5’→3’):cgaccaccttgatcttcatgctgcta)为内参,按表6配置反应体系并在荧光定量pcr仪中运行表7设置的反应程序,进行q-pcr反应。
表6.q-pcr的反应体系
表7.q-pcr的反应程序
经过q-pcr检测,我们发现gapdh目的片段得到极大富集(如图3所示)。当以水稻基因组dna为模板,进行q-pcr扩增时,内参基因actin与已知序列gapdh目的片段相对含量的差异倍数较小(二者相对含量相差在1倍以内);当以第一轮pcr的纯化产物为模板,进行q-pcr时,相比于文库中actin含量,gapdh目的片段含量得到大量富集(二者相对含量相差在3900倍以上);再接着以第二轮pcr的纯化产物为模板,进行q-pcr时,相比于文库中actin含量,gapdh目的片段含量得到极大富集(二者相对含量相差在7.8×108倍以上)。由此可知,已知序列gapdh片段的侧翼序列在两轮pcr中得到了极大富集。
实施例2通过sanger测序验证水稻tos17的侧翼序列可以被gps-seq分离
2.1转座酶打断水稻基因组(反应体系及反应程序同实例1.1)
2.2磁珠法纯化转座酶复合体打断后的产物(具体步骤见实例1.2)
2.3利用gps-seq扩增tos17的侧翼序列
按照实验要求,设计扩增tos17的侧翼序列的特异性引物,pcr扩增所需引物序列及引物结合位置信息如表8:
表8.pcr扩增tos17所需引物序列及引物结合位置信息
注:加粗斜体碱基为illumina测序接头p5的3’端碱基序列。
按表9、表10成分在灭菌的pcr管中配置两轮pcr的反应体系并在pcr仪中运行表11设置的反应程序,扩增tos17的侧翼序列。
表9.第一轮pcr反应体系
注:template1为水稻基因组dna经转座酶打断、加接头,纯化后的产物。
表10.第二轮pcr反应体系
注:template2为第一轮pcr的50μl产物,取2μl稀释50倍后的稀释液。
表11.第一轮与第二轮pcr反应程序
2.4gps-seq扩增的pcr产物纯化(具体步骤见实例1.4)
2.5pcr产物纯化后末端加“a”反应
按表12成分在灭菌的pcr管中配置pcr产物末端加“a”体系,并将反应管置于pcr仪中,运行如下反应程序:105℃热盖,30℃30min,16℃hold。
表12.pcr产物末端加“a”反应体系
2.6末端加“a”后的反应产物再次纯化
将上述反应产物补水至50μl,再次用1.8×beads纯化,纯化步骤与浓度测定方法见实例1.2。
2.7末端加“a”后的纯化产物与t载体连接
按表13成分在灭菌的pcr管中配置片段与pmd19-t载体的连接反应体系,并将反应管置于pcr仪中,运行如下反应程序:16℃连接过夜。
表13.片段与pmd19-t载体连接体系
注:目的片段与t载体之间摩尔比介于3:1~10:1之间
2.8连接产物转化大肠杆菌dh5α及蓝白斑筛选
片段与t载体连接完成后,按如下步骤将连接产物转化到大肠杆菌体内:
①大肠杆菌dh5α从-80℃冰箱拿出,冰上解冻;
②于超净台中加入5μl连接产物,用移液枪轻轻吹打混匀,冰浴30min;
③42℃热激70s,立即放于冰上2~3min;
④于超净台中加入500μl无抗生素的lb液体培养基,180rpm,37℃复苏45min;
⑤吸取200μl菌液均匀加在含amp抗性的固体lb培养皿中(表面涂有50μliptg(50mg/ml)与13μlx-gal(20mg/ml)混合物),用无菌涂布棒涂匀,在超净台中吹干后,于37℃培养箱中,倒置培养过夜。
2.9菌液pcr验证阳性单克隆
取培养过夜的平板,挑选白色单菌落各40条接种到含有amp抗性的lb液体培养管中,摇菌3-4h(37℃140rpm),并以此为模板,以pmd19-t载体上插入位点侧翼自带引物m13(正向引物f(5’→3’):tgtaaaacgacggccagt;反向引物r(5’→3’):caggaaacagctatgacc)为引物,按表14配置反应体系并置于pcr仪中运行表15的反应程序,进行菌液pcr扩增反应。
表14.菌液pcr反应体系
表15.菌液pcr反应程序
2.10利用琼脂糖凝胶电泳检测阳性克隆中插入的tos17侧翼序列片段大小
pcr反应结束后,向反应产物中各加入2μl的6×loadingbuffer,混匀并微离心,取10μl混合液点胶检测,电泳结果如图4所示。由图可得,挑选的单克隆均为阳性,阳性率为100%。
2.11水稻内源性tos17逆转座子扩增产物的单克隆sanger测序分析
取5个无菌的2ml的ep管中,加入1ml带amp氨苄青霉素抗性的lb液体培养基,然后从鉴定的阳性菌落中挑取5个单克隆(编号为1、2、5、27、40)加入培养基,并置于37℃,140rpm摇床上培养3-4h,提取质粒,以m13f为测序引物进行sanger测序。通过ncbi的blastn比对,发现1、2、5、27、40的测序序列绝大部分比对pmd19-t上,剩下的序列都比对到水稻原始拷贝的tos17序列及其右端的侧翼序列上。所以,通过ta克隆的sanger测序可得,经过两轮pcr后,水稻中tos17的侧翼序列可以得到有效分离与富集。
实施例3利用gps-seq和高通量测序同时分离水稻功能获得性突变体中多个dna元件的左右两端的侧翼序列
本发明中所用的水稻突变体是超表达水稻均一化全长cdna(fl-cdna)文库的水稻功能获得性突变体;由于创建获得性突变体的转基因过程也会激活水稻内源性tos17逆转座子的转座而造成tos17的插入。因此,该突变体库中的每个转化植株也是一个t-dna和tos17转座子插入的突变体,本实例结合gps-seq和高通量测序,同时分离水稻功能获得性突变体中t-dna、tos17元件的左右两端的侧翼序列和超表达的fl-cdna的序列。此处使用的突变体编号为k664。
3.1转座酶复合体打断水稻突变体基因组并用磁珠法纯化
利用转座酶复合体,对水稻突变体材料k664的dna,进行打断并加上gps-seq接头,并对得到的产物进行磁珠纯化,具体步骤请参见1.2。具体反应体系配置如下:
表16.转座酶打断加接头反应体系
将配置好的反应体系置于pcr管中运行如下反应程序:105℃热盖,55℃10min,10℃hold;反应产物用1.8×beads纯化,具体步骤及浓度测定方法见实例1.2。
3.2利用gps-seq特异性扩增突变体中t-dna/tos17左右两端的侧翼序列和超表达的fl_cdna的序列
突变体中插入的t-dna序列来自于重组载体,该载体包含被超表达的fl-cdna,按表18、表19成分配置pcr反应体系并于pcr管中运行表20设置的反应程序,进行gps-seq扩增,其中两轮pcr扩增所需引物见表17。
表17.pcr扩增所需引物序列及引物结合位置信息
注:加粗斜体碱基为illumina测序接头p5的3’端碱基序列。
表18.第一轮pcr反应体系
注:template1为水稻突变体k664的基因组dna经转座酶打断、加接头,纯化后的产物。
表19.第二轮pcr反应体系
注:template2为第一轮pcr的产物取2μl,稀释50倍后的稀释液。
表20.第一轮与第二轮pcr反应程序
为了使第二轮的pcr产物适用于高通量测序,还需按表21成分配置pcr反应体系并运行表22设置的反应程序,进行第三轮pcr扩增。
表21.第三轮pcr反应体系
表22.第三轮pcr的反应程序
pcr产物用1.8×beads纯化,具体步骤见实例1.2。
3.3数据分析流程
对经过三轮pcr扩增后的产物进行二代高通量测序,测序方式为pair-end150,对测序得到的原始reads进行qc检测,并对低质量的reads进行过滤,gps-seq的数据分析流程如图5所示。
3.4突变体k664的数据分析结果
通过对数据进行如上流程及igv辅助分析,我们得出了突变体k664的t-dna/tos17的插入位置及fl_cdna对应的mrna的id号(图6,7,8,9),如下表所示:
表23.k664中分离到的t-dna、tos17的插入位点及fl_cdna的序列
注:chrxrb表示t-dna分别插入到chrx上,且分离的侧翼序列是从t-dna的右边界(rightboder)分离得到;chrxlbrb为排除水稻内源的tos17拷贝外新增的插入位置,且从tos17
的左右边界均能分离到侧翼序列。
3.5利用pcr扩增验证突变体测序数据分析结果
为了验证gps-seq技术对水稻已知序列dna片段的侧翼序列的分离是否可靠,我们设计了位于基因组中t-dna/tos17插入位点两端的引物,结合位于t-dna/tos17的边界序列的引物,以k664基因组为模板,按表25成分配置pcr反应体系并置于pcr仪中运行表26设置的反应程序,进行pcr扩增验证,其中扩增所用的引物如表24所示。
表24.鉴定t-dna、tos17插入位置所需的引物
注:t-dna3f/t-dna3r引物为t-dna插入到chr3,插入位点两端的正反引物,余下同;tos17-1f/tos17-1r引物为新增的tos17插入到chr1,为插入位点两端的正反引物,余下同。
表25.pcr扩增的反应体系
表26.pcr扩增的反应程序
3.6利用琼脂糖凝胶电泳检测pcr扩增的产物大小
pcr反应结束后,向反应产物中各加入2μl的6×loadingbuffer,混匀并微微离心,取10μl混合液点胶检测,并用1%的琼脂糖凝胶检测。
如图6,结合突变体k664的igv图上的reads与日本晴参考基因组(https://www.ncbi.nlm.nih.gov/assembly/gcf_000005425.2/)的mapping情况(过滤掉比对质量分数小于30的reads)及t-dna插入位置验证的电泳图可看出,k664在chr3上约33758484bp处左边界在参考基因组上无reads覆盖,而右边界则有大量的readsmapping到参考基因组上,即t-dna右边界有侧翼序列被分离出来;由电泳图可看出,t-dna插入位点的正向引物(t-dna-3f)与左边界引物(sp_t-dnal1)扩增无条带产生而插入位点的反向引物(t-dna-3r)与右边界引物(sp_t-dnar1)扩增则有大小正确的目的片段产生,即该处有t-dna插入,且该侧翼序列是从t-dna的右边界分离得到;k664在chr7上约16672528bp处出现与chr3上相同的情况,分析略,即实验验证结果与数据分析结果一致。本实施例中,未能从t-dna左边界分离到侧翼序列,而位于t-dna插入位点左边的基因组引物和t-dna的左边界引物也未能扩增出目标片段,说明转基因过程中发生了边界丢失或载体串联重复等复杂的转基因事件,导致t-dna左边界整合到基因组中可能不是简单的插入。
如图7及图8,结合突变体k664的igv图上的reads与参考基因组的mapping情况及tos17插入位置验证的电泳图可看出,k664在chr1上约3719696bp的地方,左右边界都有大量的readsmapping到参考基因组上,即插入位点的左右边界均有侧翼序列分离出来;由电泳图可看出,tos7插入位点的正向引物(tos17-1f)与左边界引物(sp_tos17l1)及插入位点的反向引物(tos17-1r)与右边界引物(sp_tos17r1)pcr扩增均有大小正确的目的片段产生,即该处确实有tos17的插入,且左右边界均能分离到水稻的侧翼序列,其他插入位点分析相同,即实验验证结果与数据分析结果一致,突变体k664新增的tos17分别位于chr1、chr3、chr4、chr5、chr7、chr9、chr11上。
3.7水稻突变体的超表达fl_cdna的序列分析与扩增
为了找到突变体k664的fl_cdna序列,我们将包含sp_fl_cdnal2与sp_fl_cdnar2引物的reads提取出来,分别与水稻日本晴的mrna库(https://www.ncbi.nlm.nih.gov/assembly/gcf_000005425.2/)及重组载体序列进行bwa比对,发现reads在nm_001061695.1(mrna的id号)上有较高的覆盖深度,其他地方没有或极少reads覆盖,即k664的fl_cdna可能为nm_001061695.1,具体情况如图9。
为了验证数据分析得到的fl_cdna序列是否正确,我们以水稻r498基因组dna做对照,以突变体k664为实验组,用多克隆位点两侧的引物sp_fl_cdnal2与sp_fl_cdnar2扩增插入的fl_cdna序列,反应体系配置与程序设置同3.5,扩增产物用1%的琼脂糖凝胶检测,发现水稻中r498基因组dna不能扩增出条带,而k664扩增出单一的条带,结果如图10所示。
3.8水稻突变体的超表达fl_cdna序列的测序验证
将图10扩增的片段切胶回收,进行ta克隆(切胶回收方法参见相应的protocol;ta克隆详细步骤见实例2.7),以m13f为测序引物进行测序,测序结果与数据分析得出的序列进行比对,结果如下:
由表27分析可得:k664的fl_cdna序列与nm_001061695.1(491bp)oryzasativajaponicagroupos05g0311000mrna,completecds有275个碱基匹配,多克隆位点两端为t-dna序列,中间为插入的fl_cdna序列,与数据分析预测的结果一致(具体分析见表27)。
表27.k664的fl_cdnasanger测序序列与重组载体、预测的mrna的比对结果
注:query-queryid;subject-subjectid;identify-%identify;length-alignmentlength;mis-numberofmismatches;gap-totalnumberofgaps;q_start-startofalignmentinquery;q_end-endofalignmentinquery;s_start-startofalignmentinsubject;s_end-endofalignmentinsubject;e-value-expectvalue;score-rawscore。
综上所述,开发的gps-seq技术可有效富集水稻基因组中已知序列的侧翼序列,建立的数据分析方法可快速找到tos17、t-dna的插入位置及插入的fl_cdna序列,通过实验验证,可发现该方法对插入位置及未知序列的寻找准确率几乎达到100%,是一种非常简单、高效、准确的分离已知序列dna片段侧翼序列的技术。
- 还没有人留言评论。精彩留言会获得点赞!