苯并脂(肪)环衍生物及其用途的制作方法
本发明一种苯并脂(肪)环衍生物及其用途;具体地说,涉及一种由含氮杂环基的取代基取代的苯并脂(肪)环类化合物及其用途。
背景技术:
近年来,侵袭性真菌感染呈持续增多趋势,尤其在hiv感染、器官移植、恶性肿瘤、烧伤、糖尿病、结核病、囊胞性纤维症、体内导管、创伤、肿瘤化疗、长期使用抗生素及激素类药物的患者中。其中,主要的几类致病真菌如念珠菌(candida),隐球菌(cryptococcus)、曲霉菌(aspergillus)和肺孢子虫(pneumocystis)等每年导致大约200万人患病,100万人死亡。
侵袭性真菌感染的发病率逐年上升,死亡率也一直居高不下,但是相应的抗真菌药物研发却相对滞后。从19世纪50年代两性霉素b成功上市至今,用于治疗侵袭性真菌感染的药物主要仅有四种,分别是:多烯类(两性霉素b等)、棘白菌素类(卡泊芬净等)、核酸类(5-氟胞嘧啶)和唑类(氟康唑、伊曲康唑等)。尽管这些药物使得真菌感染从预防到治疗都取得了很大进展,但其对于部分侵袭性真菌感染(如隐球菌脑膜炎等)仍然不能快速发挥显著疗效,有时给药疗程要持续数月甚至更长。更严重的是,以上几种药物除了棘白菌素类都有较严重的副作用,尤其是两性霉素b,然而棘白菌素类对隐球菌无效。同时,仅有的这几种抗真菌药物的长时间反复使用,导致了耐药菌大量出现。
因此,本领域亟待需要有一批具有抗真菌活性的结构新颖的化合物出现,为新颖抗真菌药物的研制奠定坚实的基础。
技术实现要素:
本发明的发明人经广泛且深入的研究,设计并合成了结构新颖的取代的苯并脂(肪)环类化合物。经体外抑菌活性检测发现:本发明提供的取代的苯并脂(肪)环类化合物中绝大部分具有抑制真菌活性,且部分化合物的抑制真菌活性优于现有抗真菌药物。此外,本发明提供的部分取代的苯并脂(肪)环类化合物与现有抗真菌药物联用,对耐药真菌表现出良好的协同抑制作用。因此,本发明为新颖抗真菌药物或抗真菌药物的增效剂的研制奠定坚实的基础。
本发明的一个目的在于,提供一种结构新颖的取代的苯并脂(肪)环类化合物。本发明所述的苯并脂(肪)环类化合物为式i所示化合物、其立体异构体或在药学上可接受的盐:
式i中,r1和r2分别独立选自:氢(h)、羟基(oh)或羰基(
a1为二价的5~7元的脂(肪)环基或含氧(o)的脂(肪)杂环基(所述的脂(肪)杂环基是指饱和的含氧碳环基,a1与相邻苯环为“并”的关系,下同);a2为取代的6元的至少含一个氮(n)的杂环基;n为1~5的整数;
其中,所述取代的6元至少含一个氮(n)的杂环基的取代基选自下列基团中一种或两种以上(含两种):
羟基(oh),苯基,取代苯基,c1~c3的烷基,由羟基(oh)和/或苯基取代的c1~c3的烷基,吗啉基或吡啶基;
其中,所述取代苯基的取代基选自下列基团中一种或两种以上(含两种):
卤素(f、cl、br或i),苯基(包括一价和两价的苯基),c1~c3的烷基,c1~c3的全氟烷基或c1~c3的烷氧基。
本发明另一个目的在于,提供一种组合物。所述组合物包含上述苯并脂(肪)环类化合物(式i所示化合物、其立体异构体或在药学上可接受的盐)和在药理或药学上可接受的稀释剂、载体或赋形剂等添加剂。
本发明再一个目的在于,揭示上述苯并脂(肪)环类化合物的一种用途。即式i所示化合物、其立体异构体或在药学上可接受的盐在制备白色念珠菌(c.alb.sc5314)、光滑念珠菌(c.gla.8535)或/和新生隐球菌(c.neo.h99)抑制剂中的应用;或,
式i所示化合物、其立体异构体或在药学上可接受的盐在制备抑制耐氟康唑白念珠菌(c.alb.0304103)的增效剂中的应用。
此外,本发明还有一个目的在于,提供一种制备式i所示化合物的方法。所述方法的主要步骤是:首先由式ii所示化合物与式iii或iv所示化合物反应,得到式va或vb所示化合物;然后再由式va所示化合物或vb所示化合物(vb所示化合物须先进行酯的水解成相应的羧酸)与式vi所示化合物反应,得到部分目标物(式ia所示化合物);或,
将式ia所示化合物经羰基还原反应,得到另一部分目标物(式ib所示化合物);或,
将式va或vb所示化合物分别制成式vaa或vbb所示化合物,再与式vi所示化合物反应(vbb所示化合物须先进行酯的水解成相应的羧酸),得到剩余部分目标物(式ic所示化合物)。
其中,式ii、iii、iv和vi所示化合物为已知化合物,可直接市购或按现有公开文献自制,x为f、cl或br,a1和a2及n的含义与前文所述相同,r3为c1~c3烷基,r2a为羰基
附图说明
图1.为式ic-9所示化合物和氟康唑(flc,阳性对照物)对新生隐球菌(c.neo.h99)的被膜抑制活性示意图;
图2.为式ic-9所示化合物和flc作用于新生隐球菌(c.neo.h99)的时间-生长曲线;
图3.为系统性新生隐球菌(c.neo.h99)感染小鼠模型实验结果示意图。
具体实施方式
在本发明一个优选的技术方案中,a1为二价的7元的脂(肪)环基,或二价的5~7元含氧(o)的脂(肪)杂环基,其中所含氧杂原子数为1或2;
在一个更优选的技术方案中,a1为下列基团中一种:
在本发明另一个优选的技术方案中,a2为取代的哌嗪基、哌啶基、吗啉基或二氢吡啶基,
其中,所述取代的哌嗪基、哌啶基、吗啉基或二氢吡啶基的取代基选自下列基团中一种或两种以上(含两种):
羟基(oh),苯基,取代苯基,c1~c3的烷基,由羟基(oh)和/或苯基取代的c1~c3的烷基,吗啉基或吡啶基;
所述取代苯基的取代基选自下列基团中一种或两种以上(含两种):
卤素(f、cl、br或i),苯基(包括一价和两价的苯基),c1~c3的烷基,c1~c3的全氟烷基或c1~c3的烷氧基。
在进一步优选的技术方案中,a2为取代的哌嗪基、哌啶基、吗啉基或二氢吡啶基,
其中,所述取代的哌嗪基、哌啶基、吗啉基或二氢吡啶基的取代基选自下列基团中一种或两种以上(含两种):
羟基(oh),甲基,苯基,氯取代的苯基,氟取代的苯基,甲基取代的苯基,三氟甲基取的代苯基,甲氧基取代的苯基,联苯基,萘基,吗啉基,吡啶基或
在更进一步优选的技术方案中,a2为下列取代杂环基中一种:
本发明提供的制备式i所示化合物的方法,具体包括如下步骤:
(1)在冰浴条件下,由式ii所示化合物与式iii或iv所示化合物干燥的有机溶剂(如二氯甲烷(简记为“dcm”)等)中反应,用饱和氯化钠溶液淬灭反应,分液,水相用卤代烃(如dcm等)萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,残余物(粗产物)经硅胶柱柱层析,得到式va或vb所示化合物;
式vb所示化合物与一水合氢氧化锂在由甲醇、四氢呋喃和水组成的混合溶剂中进行水解反应至少3小时,调节ph至3-4,用乙酸乙酯萃取,分液,有机相用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,得到vb所示化合物的水解产物(其可直接用于下一步反应);
(2)式va所示化合物与式vi所示化合物于干燥的非质子极性有机溶剂(如n,n-二甲基甲酰胺(简记为“dmf”)等)中在50℃~80℃反应至少12小时,加入乙酸乙酯和水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,残余物(粗产物)经硅胶柱柱层析,得到式ia所示化合物;或,
vb所示化合物的水解产物、式vi所示化合物、2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(简记为“hatu”)及三乙胺(简记为“tea”)于干燥的dmf中室温搅拌至少5小时,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,得到式ia所示化合物。
或,
将式ia所示化合物和硼氢化钠置于反应瓶中,加入干燥的甲醇,室温搅拌至少1.5小时,减压浓缩除去溶剂,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,残余物(粗产物)经硅胶柱柱层析,得到式ib所示化合物。
或,
(1)在冰浴条件下,由式ii所示化合物与式iii或iv所示化合物干燥的有机溶剂(如dcm”等)中反应,用饱和氯化钠溶液淬灭反应,分液,水相用卤代烃(如dcm等)萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,残余物(粗产物)经硅胶柱柱层析,得到式va或vb所示化合物;
(2)将式va或vb所示化合物,三乙基硅烷和干燥的(三氟乙酸,简记为“tfa”)置于反应瓶中,在有惰性气体(如氮气等)存在的条件下,于50℃~80℃搅拌至少4小时,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,残余物(粗产物)经硅胶柱柱层析,得到式vaa或vbb所示化合物;
式vbb所示化合物与一水合氢氧化锂在由甲醇、四氢呋喃和水组成的混合溶剂中进行水解反应至少3小时,调节ph至3-4,用乙酸乙酯萃取,分液,有机相用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,得到式vbb所示化合物的水解产物(其可直接用于下一步反应);
(3)式vaa所示化合物与式vi所示化合物于干燥的非质子极性有机溶剂(如dmf等)中在50℃~80℃反应至少12小时,加入乙酸乙酯和水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,残余物(粗产物)经硅胶柱柱层析,得到式ic所示化合物;或,
式vbb所示化合物的水解产物,式vi所示化合物,hatu及tea于干燥的dmf中室温搅拌至少5小时,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,得到式ic所示化合物。
以下通过实施例对本发明作进一步阐述,其目的仅在于更好理解本发明的内容。因此所举之例不限制本发明的保护范围。本文中所述的室温为15℃~25℃
实施例1~43为化合物合成实施例
实施例1
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(2,3-二氢苯并呋喃-5-基)丁烷-1-酮(式ia-1所示化合物)的制备:
(1)称取2,3-二氢苯并呋喃(200mg,1.67mmol,式ii-1所示化合物),4-氯丁酰氯(259mg,1.84mmol,式iii-1所示化合物)于反应瓶中,加入10ml干燥的dcm。在冰浴条件下,缓慢加入三氯化铝(245mg,1.84mmol),维持冰浴并搅拌1.5h,缓慢加入适量饱和氯化钠溶液淬灭反应,分液,水相用二氯甲烷萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经硅胶柱柱层析(石油醚/乙酸乙酯=10:1,体积比,下同)得到白色固体(式va-1所示化合物)320mg,收率:85.6%。
1hnmr(600mhz,cdcl3,tms)δ7.87(d,j=1.2hz,1h),7.82(dd,j=8.4,1.8hz,1h),6.81(d,j=8.4hz,1h),4.66(t,j=9.0hz,2h),3.67(t,j=6.6hz,2h),3.26(t,j=9.0hz,2h),3.11(t,j=7.2hz,2h),2.24-2.18(m,2h).
(2)称取式va-1所示化合物(200mg,0.89mmol),4-(4-氯苯基)哌啶-4-醇(283mg,1.335mmol,式vi-1所示化合物)于反应瓶中,加入15ml干燥的dmf,随后加入n,n-二异丙基乙胺(344mg,2.67mmol),加热至60℃反应16h,加入乙酸乙酯和水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经硅胶柱柱层析(二氯甲烷/甲醇=100:2)得到白色固体(式ia-1所示化合物)160mg,收率:44.9%。
1hnmr(600mhz,dmso-d6,tms)δ7.88(s,1h),7.82(d,j=8.4hz,1h),7.41(d,j=8.4hz,2h),7.34(d,j=8.4hz,2h),6.86(d,j=8.4hz,1h),4.82(s,1h),4.63(t,j=8.4hz,2h),3.23(t,j=9.0hz,2h),2.91(t,j=7.2hz,2h),2.59(d,j=9.6hz,2h),2.38-2.28(m,4h),1.84-1.71(m,4h),1.50(d,j=12.2hz,2h).
ms(esi):m/z[m+h]+:400.29。
实施例2
4-(4-(4-氯苯基)-4-羟基哌啶-1-基)-1-色满-6-基丁烷-1-酮(式ia-2所示化合物)的制备:
除以色满替换实施例1中2,3-二氢苯并呋喃外,其余步骤及所用试剂与实施例1相同,得到白色固体(式ia-2所示化合物),50mg,收率:47.7%。
1hnmr(600mhz,dmso-d6,tms)δ7.74(s,1h),7.72(dd,j=8.4,1.8hz,1h),7.43(d,j=7.8hz,2h),7.35(d,j=6.6hz,2h),6.83(d,j=8.4hz,1h),4.85(s,1h),4.26-4.12(m,2h),2.93(s,2h),2.80(t,j=6.6hz,2h),2.73-2.52(m,2h),2.49-2.17(s,4h),1.97-1.92(m,2h),1.90-1.68(m,4h),1.62-1.43(s,2h).
ms(esi):m/z[m+h]+:414.30。
实施例3
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(2,3-二氢苯并[b][1,4]二氧基-6-基)丁烷-1-酮(式ia-3所示化合物)的制备:
除以2,3-二氢苯并[b][1,4]二恶嗪替换实施例1中2,3-二氢苯并呋喃外,其余步骤及所用试剂与实施例1相同,得到白色固体(式ia-3所示化合物),57mg,收率:42.4%。
1hnmr(600mhz,dmso-d6,tms)δ7.53(dd,j=8.4,1.8hz,1h),7.50(d,j=1.8hz,1h),7.44(d,j=7.8hz,2h),7.36(d,j=7.8hz,2h),7.00(d,j=8.4hz,1h),4.84(s,1h),4.36-4.32(m,2h),4.32-4.28(m,2h),2.92(s,2h),2.60(s,2h),2.45-2.22(m,4h),1.93-1.64(m,4h),1.50(s,2h).
ms(esi):m/z[m+h]+:416.32。
实施例4
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(3,4-二氢-2h-苯并[b][1,4]二氧基-7-基)丁烷-1-酮(式ia-4所示化合物)的制备:
除以3,4-二氢-2h-苯并[b][1]二氧杂环庚三烯替换实施例1中2,3-二氢苯并呋喃外,其余步骤及所用试剂与实施例1相同,得到白色固体(式ia-4所示化合物),75mg,收率:39.4%。
1hnmr(600mhz,dmso-d6,tms)δ7.57(dd,j=8.4,2.4hz,1h),7.54(d,j=2.4hz,1h),7.38(d,j=8.4hz,2h),7.32(d,j=7.8hz,2h),7.04(d,j=8.4hz,1h),4.80(s,1h),4.21(t,j=5.4hz,2h),4.16(t,j=5.4hz,2h),2.89(s,2h),2.55(s,2h),2.39-2.22(m,4h),2.16-2.11(m,2h),1.80(s,2h),1.69(s,2h),1.47(s,2h).
ms(esi):m/z[m+h]+:430.33。
实施例5
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(2,3-二氢苯并呋喃-5-基)丁烷-1-酮(式ia-5所示化合物)的制备:
(1)2,3,4,5-四氢-1-苯并[b]氧杂环庚三烯(式ii-2所示化合物)的制备:
称取2-溴苯酚(519mg,3.00mmol),4-溴正丁醇(459mg,3.00mmol)及三苯基膦(ph3p,943mg,3.60mmol)于反应瓶中,加入20ml无水的四氢呋喃(thf)。氮气保护下于0℃缓慢滴加偶氮二甲酸二乙酯(dead,783mg,4.5mmol),滴加完毕之后自然升温至室温并搅拌过夜,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(石油醚/乙酸乙酯=30:1)得到无色油状物(1-溴-2-(4-溴丁氧基)苯(式m所示化合物),简记为“中间体m”),480mg,收率:52.3%。
1hnmr(600mhz,cdcl3,tms)δ7.53(dd,j=7.8,1.8hz,1h),7.26-7.22(m,1h),6.88(dd,j=8.4,1.2hz,1h),6.83(td,j=7.8,1.8hz,1h),4.06(t,j=6.0hz,2h),3.53(t,j=6.6hz,2h),2.17-2.10(m,2h),2.02-1.96(m,2h)。
称取中间体m(200mg,0.65mmol)于反应瓶中,加入10ml无水的四氢呋喃(thf)。氮气保护下于-78℃缓慢滴加正丁基锂(n-buli,2.4m的正己烷溶液,0.33ml,0.78mmol),滴加完毕之后维持-78℃继续搅拌1.5h,加入适量饱和氯化铵溶液淬灭反应,加入乙酸乙酯稀释,分液,水相用乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(石油醚100%)得到无色油状物(式ii-2所示化合物)58mg,收率:60.9%。
(2)除以式ii-2所示化合物替换实施例1中2,3-二氢苯并呋喃外,其余步骤及所用试剂与实施例1相同,得到白色固体(式ia-5所示化合物),58mg,收率:44.2%。
1hnmr(600mhz,dmso-d6,tms)δ7.82(d,j=1.8hz,1h),7.75(dd,j=8.4,1.8hz,1h),7.46-7.35(m,2h),7.41-7.28(m,2h),7.01(d,j=7.8hz,1h),4.81(s,1h),4.05-3.91(m,2h),3.07-2.86(m,2h),2.85-2.77(m,2h),2.70-2.49(m,2h),2.45-2.11(m,4h),1.94-1.36(m,10h).
ms(esi):m/z[m+h]+:428.35。
实施例6
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丙烷-1-酮(式ia-6所示化合物)的制备:
(1)称取6,7,8,9四氢-5h-苯并[7]轮烯-5-酮(200mg,1.25mmol),三乙基硅烷(434mg,3.75mmol)于反应瓶中,加入10ml干燥的三氟乙酸(tfa)。氮气保护下于60℃搅拌4h,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(100%石油醚)得到无色油状物(式ii-3所示化合物)150mg,收率:82.2%。
1hnmr(600mhz,cdcl3,tms)δ7.87(d,j=1.2hz,1h),7.82(dd,j=8.4,1.8hz,1h),6.81(d,j=8.4hz,1h),4.66(t,j=9.0hz,2h),3.67(t,j=6.6hz,2h),3.26(t,j=9.0hz,2h),3.11(t,j=7.2hz,2h),2.24-2.18(m,2h).
(2)称取式ii-3所示化合物(120mg,0.82mmol),3-氯丙酰氯(113mg,0.90mmol)于反应瓶中,加入10ml干燥的dcm。冰浴下条件下,缓慢加入三氯化铝(120mg,0.90mmol),维持冰浴并搅拌1.5h,缓慢加入适量饱和氯化钠溶液淬灭反应,分液,水相用二氯甲烷萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(石油醚/乙酸乙酯=10:1)得到白色固体(式va-2所示化合物)157mg,收率:80.9%。
1hnmr(300mhz,cdcl3,tms)δ7.73-7.64(m,2h),7.19(d,j=7.5hz,1h),3.92(t,j=6.9hz,2h),3.43(t,j=6.9hz,2h),2.91-2.80(m,4h),1.91-1.79(m,2h),1.72-1.59(m,4h).
(3)称取式va-2所示化合物(70mg,0.30mmol),4-(4-氯苯基)哌啶-4-醇(95mg,0.45mmol,式vi-1所示化合物)于反应瓶中,加入10ml干燥的dmf,随后加入n,n-二异丙基乙胺(116mg,0.90mmol),加热至60℃反应16h,加入乙酸乙酯和水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(二氯甲烷/甲醇=100:2)得到白色固体(式ia-6所示化合物)53mg,收率:43.1%。
1hnmr(600mhz,dmso-d6,tms)δ7.73(d,j=1.8hz,1h),7.70(dd,j=7.8,1.8hz,1h),7.48(d,j=9.0hz,2h),7.35(d,j=8.4hz,2h),7.25(d,j=7.8hz,1h),4.87(s,1h),3.16(t,j=7.8hz,2h),2.87-2.80(m,4h),2.73-2.66(m,4h),2.43(t,j=10.8hz,2h),1.90-1.76(m,4h),1.62-1.52(m,6h).
ms(esi):m/z[m+h]+:412.41。
实施例7
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁烷-1-酮(式ia-7所示化合物)的制备:
除以4-氯丁酰氯替换实施例6中3-氯丙酰氯外,其余步骤及所用试剂与实施例6相同,得到白色固体(式ia-7所示化合物),45mg,收率:42.7%。
1hnmr(600mhz,dmso-d6,tms)δ7.70(s,1h),7.68(d,j=7.8,1.8hz,,1h),7.34(d,j=8.4hz,2h),7.31(d,j=8.4hz,2h),7.24(d,j=7.8hz,1h),4.80(s,1h),2.92(s,2h),2.85-2.77(m,4h),2.64-2.50(m,2h),2.42-2.19(m,4h),1.85-1.73(m,4h),1.72-1.59(m,2h),1.59-1.48(m,4h),1.48-1.37(m,2h).
ms(esi):m/z[m+h]+:426.36。
实施例8
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)戊烷-1-酮(式ia-8所示化合物)的制备:
除以5-氯戊酰氯替换实施例6中3-氯丙酰氯外,其余步骤及所用试剂与实施例6相同,得到白色固体(式ia-8所示化合物),46mg,收率:46.7%。
1hnmr(600mhz,dmso-d6,tms)δ7.72-7.67(m,2h),7.46(d,j=8.4hz,2h),7.35(d,j=8.4hz,2h),7.25(d,j=7.7hz,1h),4.85(s,1h),3.05-2.95(m,2h),2.88-2.79(m,4h),2.68-2.55(m,2h),2.39-2.22(m,4h),1.91-1.75(m,4h),1.66-1.60(m,2h),1.60-1.42(m,8h).
ms(esi):m/z[m+h]+:440.45。
实施例9
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)己烷-1-酮(式ia-9所示化合物)的制备:
除以6-溴己酰氯替换实施例6中3-氯丙酰氯外,其余步骤及所用试剂与实施例6相同,得到白色固体(式ia-9所示化合物),55mg,收率:40.2%。
1hnmr(600mhz,dmso-d6,tms)δ7.70(s,1h),7.70-7.67(m,1h),7.48(d,j=8.4hz,2h),7.36(d,j=8.4hz,2h),7.24(d,j=7.8hz,1h),4.90(s,1h),2.98(t,j=7.2hz,2h),2.86-2.80(m,4h),2.78-2.58(m,2h),2.47-2.21(m,4h),1.95-1.84(m,2h),1.83-1.77(m,2h),1.65-1.60(m,2h),1.60-1.53(m,6h),1.53-1.43(s,2h),1.37-1.30(m,2h).
ms(esi):m/z[m+h]+:454.46。
实施例10
4-[4-(4-氯苯基)-4-羟基哌啶-1-基]-1-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)庚烷-1-酮(式ia-10所示化合物)的制备:
除以7-溴庚酰氯替换实施例6中3-氯丙酰氯外,其余步骤及所用试剂与实施例6相同,得到白色固体(式ia-10所示化合物),35mg,收率:35.1%。
1hnmr(600mhz,dmso-d6,tms)δ7.69(d,j=1.2hz,1h),,7.68(dd,j=7.8,1.8hz,1h),7.48(d,j=8.4hz,2h),7.36(d,j=7.8hz,2h),7.24(d,j=7.8hz,1h),4.91(s,1h),2.97(t,j=7.2hz,2h),2.86-2.79(m,4h),2.78-2.59(m,2h),2.48-2.13(m,4h),2.00-1.85(m,2h),1.83-1.77(m,2h),1.65-1.53(m,8h),1.52-1.41(m,2h),1.37-1.30(m,4h).
ms(esi):m/z[m+h]+:468.46。
实施例11
(4-(4-氯苯基)-4-羟基哌啶-1-基)-4-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)-1,4-丁二酮(式ia-11所示化合物)的制备:
(1)式ii-3所示化合物(250mg,1.71mmol),4-氯-4-氧代丁酸甲酯(282mg,1.88mmol,式iii-3所示化合物)于反应瓶中,加入10ml干燥的dcm。冰浴条件下,缓慢加入三氯化铝(250mg,1.88mmol),维持冰浴并搅拌1.5h,缓慢加入适量饱和氯化钠溶液淬灭反应,分液,水相用二氯甲烷萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(石油醚/乙酸乙酯=10:1)得到淡黄色固体(式vb-1所示化合物)320mg,收率:71.9%。
1hnmr(600mhz,cdcl3,tms)δ7.73-7.70(m,2h),7.18(d,j=7.8hz,1h),3.70(s,3h),3.30(t,j=7.2hz,2h),2.87-2.82(m,4h),2.76(t,j=6.6hz,2h),1.88-1.82(m,2h),1.68-1.61(m,4h)。
(2)称取式vb-1所示化合物(140mg,0.54mmol),一水合氢氧化锂(34mg,0.81mmol)于反应瓶中,加入5ml/5ml/2.5ml的四氢呋喃/甲醇/水(thf/meoh/h2o)混合物作为反应溶剂,室温搅拌3h,用1n的hcl调节ph至3-4,用乙酸乙酯萃取,分液,有机相用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,得到白色固体(式vb-1-oh所示化合物)120mg,收率:90.2%,直接用于下一步反应反应。
1hnmr(300mhz,dmso-d6,tms)δ12.12(s,1h),7.75-7.66(m,2h),7.26(d,j=7.5hz,1h),3.21(t,j=6.3hz,2h),2.88-2.79(m,4h),2.56(t,j=6.3hz,2h),1.88-1.72(m,2h),1.64-1.51(m,4h).
(3)称取式vb-1-oh所示化合物(120mg,0.49mmol),4-(4-氯苯基)-哌啶-4-醇(125mg,0.59mmol,式vi-1所示化合物),hatu(281mg,0.74mmol)及tea(148mg,1.47mmol)于反应瓶中,加入15ml干燥的dmf作为反应溶剂,室温搅拌5h,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,得到淡黄色色固体(式ia-11所示化合物)165mg,收率:76.7%。
1hnmr(600mhz,dmso-d6,tms)δ7.73(s,1h),7.71(dd,j=7.2,1.8hz,1h),7.52-7.50(m,2h),7.40-7.37(m,2h),7.26(d,j=7.8hz,1h),5.24(s,1h),4.28(d,j=13.2hz,1h),3.86(d,j=13.2hz,1h),3.44(td,j=13.2,2.4hz,1h),3.27-3.13(m,2h),2.94(td,j=12.6,2.4hz,1h),2.87-2.81(m,4h),2.75-2.70(m,2h),1.95(td,j=13.2,4.2hz,1h),1.85-1.77(m,2h),1.72(td,j=13.2,4.8hz,1h),1.64(dd,j=13.2,1.8hz,1h),1.62-1.52(m,5h).
ms(esi):m/z[m+h]+:440.53。
实施例12
4-(4-(4-氯苯基)-3,6-二氢吡啶-1(2h)-基)-1-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁烷-1-酮(式ia-12所示化合物)的制备:
(1)称取4-(4-氯苯基)-哌啶-4-醇(300mg,1.42mmol,式vi-1所示化合物),三乙基硅烷(434mg,4.26mmol)于反应瓶中,加入10ml干燥的tfa,氮气保护下于60℃搅拌4h,减压浓缩出去大部分溶剂,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(二氯甲烷/甲醇=100:5)得到淡黄色固体(4-(4-氯苯基)-1,2,3,6-四氢吡啶,式vi-2所示化合物)205mg,收率:74.8%。
1hnmr(600mhz,dmso-d6,tms)δ7.45(d,j=9.0hz,2h),7.39(d,j=9.0hz,2h),6.24(s,1h),3.47(d,j=2.4hz,2h),3.03(t,j=5.4hz,2h),2.41(d,j=1.8hz,2h).
(2)除以4-氯丁酰氯替换实施例6中3-氯丙酰氯,及式vi-2所示化合物替换实施例6中式vi-1所示化合物外,其余步骤及所用试剂与实施例6相同,得到白色固体(式ia-12所示化合物),72mg,收率:34.0%。
1hnmr(600mhz,dmso-d6,tms)δ7.69(s,1h),7.68(dd,j=7.8,1.8hz,1h),7.43(d,j=8.4hz,2h),7.37(d,j=9.0hz,2h),7.23(d,j=7.8hz,1h),6.18(s,1h),3.06(s,2h),3.00(t,j=6.6hz,2h),2.84-2.78(m,4h),2.63-2.57(m,2h),2.47-2.42(m,2h),2.43-2.37(m,2h),1.87-1.76(m,4h),1.59-1.50(m,4h).
ms(esi):m/z[m+h]+:408.43。
实施例13
4-(4-(4-氯苯基)-哌嗪-1-基)-1-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁烷-1-酮(式ia-13所示化合物)的制备:
除以4-氯丁酰氯替换实施例6中3-氯丙酰氯,及式vi-3(1-(4-氯苯基)哌嗪)所示化合物替换实施例6中式vi-1所示化合物外,其余步骤及所用试剂与实施例6相同,得到白色固体(式ia-13所示化合物),95mg,收率:49.1%。
1hnmr(600mhz,dmso,tms)δ7.72-7.66(m,2h),7.26-7.18(m,3h),6.91(d,j=9.0hz,2h),3.16-3.00(m,4h),3.02-2.95(m,2h),2.86-2.78(m,4h),2.50-2.42(m,4h),2.43-2.27(m,2h),1.88-1.74(m,4h),1.55(s,4h).
ms(esi):m/z[m+h]+:411.47。
实施例14
4-(4-苯基哌嗪-1-基)-1-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁烷-1-酮(式ia-14所示化合物)的制备:
除以4-氯丁酰氯替换实施例6中3-氯丙酰氯,及式vi-4所示化合物(1-苯基哌嗪)替换实施例6中式vi-1所示化合物外,其余步骤及所用试剂与实施例6相同,得到白色固体(式ia-14所示化合物),124mg,收率:44.2%。
1hnmr(600mhz,dmso-d6,tms)δ7.73-7.66(m,2h),7.24(d,j=7.8hz,1h),7.20(t,j=7.8hz,2h),6.90(d,j=7.8hz,2h),6.76(t,j=7.2hz,1h),3.12-3.01(m,4h),2.99(t,j=6.6hz,2h),2.87-2.77(m,4h),2.47-2.29(m,2h),1.89-1.73(m,4h),1.55(s,4h).
ms(esi):m/z[m+h]+:377.50。
实施例15
4-(4-氯苯基)-1-(3-羟基-3-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)丙基)哌啶-4-醇(式ib-1所示化合物)的制备:
称取式ia-6所示化合物(70mg,0.17mmol),硼氢化钠(32mg,0.8mmol)于反应瓶中,加入10ml干燥的甲醇,室温搅拌1.5h,减压浓缩除去溶剂,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(二氯甲烷/甲醇=100:5)得到白色固体(式ib-1所示化合物)52mg,收率:74.3%。
1hnmr(600mhz,dmso-d6,tms)δ7.49(d,j=8.4hz,2h),7.36(d,j=8.4hz,2h),7.06(s,1h),7.05-7.00(m,2h),5.47(s,1h),4.94(s,1h),4.58-4.54(m,1h),2.77-2.70(m,6h),2.50-2.29(m,4h),1.96-1.85(m,2h),1.82-1.71(m,4h),1.64-1.50(m,6h).
ms(esi):m/z[m+h]+:414.34。
实施例16
4-(4-氯苯基)-1-(4-羟基-4-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ib-2所示化合物)的制备:
除以式ia-7所示化合物替换实施例15中式ia-6所示化合物外,其余步骤及试剂与实施例15相同,得到白色固体(式ib-2所示化合物)59mg,收率:81.9%。
1hnmr(600mhz,dmso-d6,tms)δ7.48(d,j=8.4hz,2h),7.36(d,j=8.4hz,2h),7.05(s,1h),7.04-6.98(m,2h),5.41(s,1h),4.96(s,1h),4.45(s,1h),2.85-2.59(m,6h),2.49-2.16(m,4h),2.01-1.83(m,2h),1.82-1.74(m,2h),1.66-1.49(m,9h),1.44(s,1h).
ms(esi):m/z[m+h]+:428.38。
实施例17
4-(4-氯苯基)-1-(5-羟基-5-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)戊基)哌啶-4-醇(式ib-3所示化合物)的制备:
除以式ia-8所示化合物替换实施例15中式ia-6所示化合物外,其余步骤及试剂与实施例15相同,得到白色固体(式ib-3所示化合物)57mg,收率:76.0%。
1hnmr(600mhz,dmso-d6,tms)δ7.46(d,j=7.8hz,2h),7.32(d,j=7.8hz,2h),7.05-6.92(m,3h),4.96(s,1h),4.82(s,1h),4.40(s,1h),2.70(s,4h),2.58(d,j=9.6hz,2h),2.28(t,j=10.8hz,2h),2.23(t,j=6.6hz,2h),1.84(t,j=12.6hz,2h),1.75(s,2h),1.64-1.45(m,8h),1.40(s,2h),1.32(s,2h).
ms(esi):m/z[m+h]+:442.39。
实施例18
4-(4-氯苯基)-1-(6-羟基-6-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)己基)哌啶-4-醇(式ib-4所示化合物)的制备:
除以式ia-9所示化合物替换实施例15中式ia-6所示化合物外,其余步骤及试剂与实施例15相同,得到白色固体(式ib-4所示化合物)61mg,收率:79.2%。
1hnmr(600mhz,dmso-d6,tms)δ7.48(d,j=8.4hz,2h),7.34(d,j=8.4hz,2h),7.07-6.95(m,3h),4.95(s,1h),4.84(s,1h),4.41(s,1h),2.78-2.68(m,4h),2.60(d,j=9.6hz,2h),2.30(t,j=11.4hz,2h),2.25(t,j=7.2hz,2h),1.91-1.82(m,2h),1.78(s,2h),1.64-1.46(m,8h),1.44-1.35(m,2h),1.31-1.18(m,4h).
ms(esi):m/z[m+h]+:456.39。
实施例19
4-(4-氯苯基)-1-(7-羟基-7-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)庚基)哌啶-4-醇(式ib-5所示化合物)的制备:
除以式ia-10所示化合物替换实施例15中式ia-6所示化合物外,其余步骤及试剂与实施例15相同,得到白色固体(式ib-5所示化合物的盐酸盐)60mg,收率:75.9%。
1hnmr(600mhz,dmso-d6,tms)δ10.32(s,1h),7.48(d,j=8.4hz,2h),7.43(d,j=8.4hz,2h),7.05-6.97(m,3h),5.55(s,1h),4.99(d,j=4.2hz,1h),4.46-4.37(m,1h),3.16(s,2h),3.03(s,2h),2.77-2.68(m,4h),2.34(s,2h),1.82-1.72(m,4h),1.68(s,2h),1.63-1.49(m,6h),1.41-1.19(m,8h).
ms(esi):m/z[m+h]+:470.40。
实施例20
1-(4-4(4-氯苯基)-4-羟基哌啶-1-基)-4-羟基-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)丁-1-酮(式ib-6所示化合物)的制备:
除以式ia-11所示化合物替换实施例15中式ia-6所示化合物外,其余步骤及试剂与实施例15相同,得到白色固体(式ib-6所示化合物)51mg,收率:68.0%。
1hnmr(600mhz,dmso-d6,tms)δ7.49(d,j=8.4hz,2h),7.37(d,j=7.8hz,2h),7.07-6.98(m,3h),5.22(s,1h),5.13(t,j=4.8hz,1h),4.51-4.45(m,1h),4.33(d,j=13.2hz,1h),3.68(d,j=13.2hz,1h),3.37(d,j=13.2hz,1h),2.90(t,j=12.0hz,1h),2.73(s,4h),2.40-2.34(m,2h),1.89-1.67(m,6h),1.62-1.49(m,6h).
ms(esi):m/z[m+h]+:442.38。
实施例21
(4-4(4-氯苯基)哌嗪-1-基)-1-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)丁-1-醇(式ib-7所示化合物)的制备:
除以式ia-13所示化合物替换实施例15中式ia-6所示化合物外,其余步骤及试剂与实施例15相同,得到白色固体(式ib-7所示化合物)43mg,收率:61.4%。
1hnmr(600mhz,dmso-d6,tms)δ7.21(d,j=9.0hz,2h),7.04(s,1h),7.02(d,j=7.8hz,1h),6.99(d,j=7.8hz,1h),6.92(d,j=9.0hz,2h),5.15(s,1h),4.47-4.41(m,1h),3.10(s,4h),2.76-2.70(m,4h),2.50-2.40(m,4h),2.31(s,2h),1.81-1.75(m,2h),1.66-1.46(m,8h).
ms(esi):m/z[m+h]+:413.37。
实施例22
4-(4-苯基哌嗪-1基)-1-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)丁-1-醇(式ib-8所示化合物)的制备:
除以式ia-14所示化合物替换实施例15中式ia-6所示化合物外,其余步骤及试剂与实施例15相同,得到白色固体(式ib-8所示化合物)41mg,收率:64.1%。
1hnmr(600mhz,dmso-d6,tms)δ7.20(t,j=7.8hz,2h),7.06-6.97(m,4h),6.91(d,j=8.4hz,2h),6.76(t,j=7.2hz,1h),5.18(s,1h),4.48-4.43(m,1h),3.11(s,4h),2.78-2.68(m,6h),2.33(s,2h),1.82-1.75(m,2h),1.66-1.46(m,10h).
ms(esi):m/z[m+h]+:379.40。
实施例23
4-(4-氯苯基)-1-(3-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)丙基)哌啶-4-醇(式ic-1所示化合物)的制备:
(1)称取式ii-3所示化合物(120mg,0.82mmol),3-氯丙酰氯(113mg,0.90mmol)于反应瓶中,加入10ml干燥的dcm。冰浴下条件下,缓慢加入三氯化铝(120mg,0.90mmol),维持冰浴并搅拌1.5h,缓慢加入适量饱和氯化钠溶液淬灭反应,分液,水相用二氯甲烷萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(石油醚/乙酸乙酯=10:1)得到白色固体(式va-2所示化合物)157mg,收率:80.9%。
1hnmr(300mhz,cdcl3,tms)δ7.73-7.64(m,2h),7.19(d,j=7.5hz,1h),3.92(t,j=6.9hz,2h),3.43(t,j=6.9hz,2h),2.91-2.80(m,4h),1.91-1.79(m,2h),1.72-1.59(m,4h).
(2)称取式va-2所示化合物(180mg,0.76mmol),三乙基硅烷(264mg,2.28mmol)于反应瓶中,加入10ml干燥的tfa。氮气保护下,于60℃搅拌4h,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(100%石油醚)得到无色油状物(式vaa-1所示化合物)142mg,收率:83.9%。
1hnmr(300mhz,cdcl3)δ7.10(d,j=7.8hz,1h),7.04-6.95(m,2h),3.62(t,j=6.3hz,2h),2.85(m,4h),2.66(t,j=7.2hz,2h),1.88-1.70(m,8h).
(3)称取式vaa-1所示化合物(142mg,0.64mmol),4-(4-氯苯基)哌啶-4-醇(203mg,0.96mmol,式vi-1所示化合物)于反应瓶中,加入10ml干燥的dmf,随后加入n,n-二异丙基乙胺(248mg,1.92mmol),加热至60℃反应16h,加入乙酸乙酯和水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(二氯甲烷/甲醇=100:2)得到白色固体(式ic-1所示化合物的盐酸盐)97mg,收率:38.1%。
1hnmr(600mhz,dmso-d6,tms)δ10.34(s,1h),7.48(d,j=8.4hz,2h),7.43(d,j=8.4hz,2h),7.03(d,j=7.2hz,1h),6.98(s,1h),6.93(d,j=7.2hz,1h),5.55(s,1h),3.42(s,2h),3.20(s,2h),3.11(s,2h),2.73(m,4h),2.57(t,j=7.2hz,2h),2.41-2.30(m,2h),2.02(s,2h),1.82-1.74(m,4h),1.55(s,4h).
ms(esi):m/z[m+h]+:398.77。
实施例24
4-(4-氯苯基)-1-(4-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ic-2所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-2所示化合物的盐酸盐)89mg,收率:32.7%。
1hnmr(600mhz,dmso-d6,tms)δ10.53(s,1h),7.47(d,j=9.0hz,2h),7.43(d,j=9.0hz,2h),7.00(d,j=7.2hz,1h),6.95(s,1h),6.90(d,j=7.2hz,1h),5.56(s,1h),3.42-3.35(m,2h),3.22-3.14(m,2h),3.14-3.06(m,2h),2.74-2.681(m,4h),2.57-2.51(m,2h),2.44-2.33(s,2h),1.82-1.67(m,6h),1.63-1.50(m,6h).
ms(esi):m/z[m+h]+:412.77。
实施例25
4-(4-氯苯基)-1-(5-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)戊基)哌啶-4-醇(式ic-3所示化合物)的制备:
除5-氯戊酰氯替换实施例23中3-氯丙酰氯外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-3所示化合物)93mg,收率:36.4%。
1hnmr(600mhz,dmso-d6,tms)δ7.48(d,j=9.0hz,2h),7.35(d,j=9.0hz,2h),6.98(d,j=7.2hz,1h),6.91(s,1h),6.86(d,j=7.2hz,1h),4.83(s,1h),2.73-2.67(m,4h),2.64-2.58(m,2h),2.49-2.46(m,2h),2.35-2.24(m,4h),1.90-1.82(m,2h),1.80-1.73(m,2h),1.58-1.50(m,8h),1.49-1.42(m,2h),1.32-1.26(m,2h).
ms(esi):m/z[m+h]+:426.78。
实施例26
4-(4-氯苯基)-1-(6-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)己基)哌啶-4-醇(式ic-4所示化合物)的制备:
除6-溴己酰氯替换实施例23中3-氯丙酰氯外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-4所示化合物)80mg,收率:32.5%。
1hnmr(600mhz,dmso-d6,tms)δ7.48(d,j=8.4hz,2h),7.36(d,j=7.8hz,2h),6.98(d,j=7.2hz,1h),6.91(s,1h),6.86(d,j=7.8hz,1h),5.15-4.65(s,1h),2.73-2.67(m,4h),2.53-2.44(m,6h),2.40-2.20(m,2h),1.91(s,2h),1.80-1.73(m,2h),1.63-1.41(m,10h),1.33-1.27(s,4h).
ms(esi):m/z[m+h]+:440.76。
实施例27
4-(4-氯苯基)-1-(7-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)庚基)哌啶-4-醇(式ic-5所示化合物)的制备:
除7-溴庚酰氯替换实施例23中3-氯丙酰氯外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-5所示化合物的盐酸盐)71mg,收率:29.0%。
1hnmr(600mhz,dmso-d6,tms)δ10.06(s,1h),7.40(d,j=8.4hz,2h),7.26(d,j=8.4hz,2h),7.01(d,j=7.8hz,1h),6.95(s,1h),6.91(d,j=7.2hz,1h),3.53(d,j=11.6hz,2h),3.10-3.04(m,2h),3.03-2.94(m,2h),2.87-2.79(m,1h),2.75-2.69(m,4h),2.54(t,j=7.8hz,2h),2.03-1.91(m,4h),1.81-1.75(m,2h),1.75-1.68(m,2h),1.63-1.50(m,6h).
ms(esi):m/z[m+h]+:456.76。
实施例28
1-(4-(4-氯苯基)-4-羟基哌啶-1-基)-4-(6,7,8,9,-四氢-5h-苯并[7]轮烯-2-基)-丁-1-酮(式ic-6所示化合物)的制备:
(1)称取式vb-1所示化合物(参见实施例11)(198mg,0.76mmol),三乙基硅烷(264mg,2.28mmol)于反应瓶中,加入10ml干燥的tfa。氮气保护下于60℃搅拌4h,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(100%石油醚)得到无色油状物(式vbb-1所示化合物)163mg,收率:87.2%。
(2)称取式vbb-1所示化合物(163mg,0.66mmol),一水合氢氧化锂(34mg,0.99mmol)于反应瓶中,加入5ml/5ml/2.5ml的四氢呋喃/甲醇/水(thf/meoh/h2o)混合物作为反应溶剂,室温搅拌3h,用1nhcl调节ph至3-4,用乙酸乙酯萃取,分液,有机相用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,得到白色固体(式vbb-1-oh所示化合物)129mg,收率:84.2%,直接用于下一步反应反应。
(3)称取式vbb-1-oh所示化合物(129mg,0.56mmol),4-(4-氯苯基)-哌啶-4-醇(142mg,0.67mmol,式vi-1所示化合物),2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu,319mg,0.84mmol)及tea(170mg,1.68mmol)于反应瓶中,加入15ml干燥的dmf作为反应溶剂,室温搅拌5h,加入适量乙酸乙酯及水稀释反应液,分液,水相用乙酸乙酯萃取,合并有机相,依次用水及饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,得到淡黄色色固体(式ic-6所示化合物)192mg,收率:80.5%。
1hnmr(600mhz,dmso-d6,tms)δ7.49(d,j=7.8hz,2h),7.37(d,j=7.8hz,2h),6.99(d,j=7.8hz,1h),6.92(s,1h),6.88(d,j=7.2hz,1h),5.22(s,1h),4.34(d,j=11.4hz,1h),3.69(d,j=13.2hz,1h),3.36(t,j=12.6hz,1h),2.94-2.87(m,1h),2.73-2.67(m,4h),2.52(t,j=7.2hz,2h),2.34(t,j=7.2hz,2h),1.85-1.69(m,6h),1.61-1.48(m,6h).
ms(esi):m/z[m+h]+:426.65。
实施例29
1-(4-氯苯基)-4-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌嗪(式ic-7所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-3所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-7所示化合物的盐酸盐)75mg,收率:28.6%。
1hnmr(600mhz,dmso-d6,tms)δ10.71(s,1h),7.40(d,j=8.4hz,2h),7.27(d,j=8.5hz,2h),7.00(d,j=7.8hz,1h),6.95(d,j=1.8hz,1h),6.91(dd,j=7.8,1.8hz,1h),3.51(d,j=11.4hz,2h),3.08-3.02(m,2h),3.02-2.94(m,2h),2.87-2.79(m,1h),2.74-2.68(m,4h),2.56-2.51(m,2h),2.14-2.04(m,2h),1.94(s,1h),1.92(s,1h),1.82-1.71(m,4h),1.62-1.50(m,6h).
ms(esi):m/z[m+h]+:397.60。
实施例30
1-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基-4-(4-(三氟甲基)苯基)哌啶-4-醇(式ic-8所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-5所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-8所示化合物的盐酸盐)125mg,收率:42.5%。
1hnmr(600mhz,dmso-d6,tms)δ10.23(s,1h),7.75(d,j=7.2hz,2h),7.69(d,j=7.8hz,2h),7.01(d,j=7.8hz,1h),6.96(s,1h),6.91(d,j=7.2hz,1h),5.70(s,1h),3.41(s,2h),3.22(s,2h),3.13(s,2h),2.76-2.67(m,4h),2.55(t,j=7.2hz,2h),2.45-2.34(m,2h),1.86-1.68(m,6h),1.65-1.49(m,6h).
hrms(esi):m/z[m+h]+:446.2672。
实施例31
4-(4-氟苯基)-1-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ic-9所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-6所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-9所示化合物的盐酸盐)107mg,收率:41.0%。。
1hnmr(600mhz,dmso-d6,tms)δ10.34(s,1h),7.54-7.46(m,2h),7.19(t,j=8.4hz,2h),7.01(d,j=7.2hz,1h),6.96(s,1h),6.91(d,j=7.8hz,1h),5.52(s,1h),3.50-3.33(m,2h),3.16(s,2h),3.08(s,2h),2.72(dd,j=10.8,6.6hz,4h),2.55(t,j=7.2hz,2h),2.40-2.30(m,2h),1.81-1.74(m,4h),1.76-1.69(m,2h),1.63-1.57(m,2h),1.58-1.50(m,4h).
hrms(esi):m/z[m+h]+:396.2707。
实施例32
4-(吡啶-3-基)-1-(4-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ic-10所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-7所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-10所示化合物的盐酸盐)75mg,收率:30.0%。
1hnmr(600mhz,dmso-d6,tms)δ10.49(s,1h),8.71(s,1h),8.49(d,j=4.2hz,1h),7.85(d,j=7.8hz,1h),7.41(dd,j=8.0,4.8hz,1h),7.01(d,j=7.8hz,1h),6.96(s,1h),6.91(dd,j=7.2,1.2hz,1h),5.73(s,1h),3.43(s,2h),3.21(s,2h),3.12(s,2h),2.75-2.69(m,4h),2.55(t,j=7.8hz,2h),2.44(t,j=12.0hz,2h),1.85(d,j=13.8hz,2h),1.81-1.71(m,4h),1.64-1.58(m,2h),1.55(s,4h).
hrms(esi):m/z[m+h]+:379.2755。
实施例33
4-甲基苯基-1-(4-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ic-11所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-8所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-11所示化合物的盐酸盐)102mg,收率:41.1%。
1hnmr(600mhz,dmso-d6,tms)δ10.12(s,1h),7.34(d,j=7.8hz,2h),7.17(d,j=7.8hz,2h),7.01(d,j=7.8hz,1h),6.96(s,1h),6.91(d,j=7.2hz,1h),5.38(s,1h),3.41-3.34(m,2h),3.24-3.14(m,2h),3.11(s,2h),2.75-2.68(m,4h),2.55(t,j=7.8hz,2h),2.35-2.28(m,2h),2.28(s,3h),1.83-1.67(m,6h),1.62-1.56(m,2h),1.58-1.49(m,4h).
hrms(esi):m/z[m+h]+:392.2956。
实施例34
4-(4-(1,1’-联苯基)-1-(4-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ic-12所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-9所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-12所示化合物的盐酸盐)100mg,收率:40.8%。
1hnmr(600mhz,dmso-d6,tms)δ9.85(s,1h),7.66(d,j=7.2hz,4h),7.55(d,j=7.8hz,2h),7.46(t,j=7.2hz,2h),7.36(t,j=7.2hz,1h),7.01(d,j=7.2hz,1h),6.96(s,1h),6.91(d,j=7.2hz,1h),5.50(s,1h),3.50-3.34(m,2h),3.27-3.01(m,4h),2.75-2.69(m,4h),2.55(t,j=7.2hz,2h),2.41-2.18(m,2h),1.89-1.65(m,6h),1.63-1.51(m,6h).
hrms(esi):m/z[m+h]+:454.3113。
实施例35
二苯基-1-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-甲醇(式ic-13所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-10所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-13所示化合物的盐酸盐)72mg,收率:50.9%。
1hnmr(600mhz,dmso-d6,tms)δ10.23(s,1h),7.75(d,j=7.2hz,2h),7.69(d,j=7.8hz,2h),7.01(d,j=7.8hz,1h),6.96(s,1h),6.91(d,j=7.2hz,1h),5.70(s,1h),3.41(s,2h),3.22(s,2h),3.13(s,2h),2.76-2.67(m,4h),2.55(t,j=7.2hz,2h),2.45-2.34(m,2h),1.86-1.68(m,6h),1.65-1.49(m,6h).
hrms(esi):m/z[m+h]+:468.3263。
实施例36
(2s,6r)-2,6-二甲基-4-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)吗啉(式ic-14所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-11所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-14所示化合物)72mg,收率:50.9%。
1hnmr(300mhz,dmso-d6,tms)δ6.98(d,j=7.5hz,1h),6.90(s,1h),6.85(d,j=7.8hz,1h),3.56-3.45(m,2h),2.76-2.61(m,6h),2.22(t,j=7.2hz,2h),1.83-1.72(m,2h),1.60-1.45(m,8h),1.44-1.34(m,2h),1.23(s,2h),1.02(d,j=6.3hz,6h).
ms(esi):m/z[m+h]+:316.44。
实施例37
1-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基-4-(4-(甲氧基苯基)哌啶-4-醇(式ic-15所示化合物)的制备:
(1)4-(4-甲氧基苯基)哌啶-4-醇(式vi-12所示化合物)的制备:
称取化合物4-甲氧基溴苯(374mg,2.00mmol)于反应瓶中,加入20ml无水thf。氮气保护下于-78℃缓慢滴加正丁基锂(n-buli,2.4m的正己烷溶液,0.88ml,2.1mmol),维持-78℃搅拌1h。然后滴加缓慢滴加n-苄基哌啶酮(416mg,2.20mmol),滴加完成后继续于-78℃搅拌2h。加入适量饱和氯化铵溶液淬灭反应,加入乙酸乙酯稀释,分液,水相用乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥,减压浓缩,粗品经柱层析(二氯甲烷:甲醇=100:2)得到黄色油状物(式n所示化合物)共295mg,收率:49.6%。
1hnmr(300mhz,dmso-d6,tms)δ7.37(d,j=8.7hz,2h),7.33(s,2h),7.32(s,2h),7.28-7.22(m,1h),6.85(d,j=9.0hz,2h),4.68(s,1h),3.72(s,3h),3.49(s,2h),2.63-2.54(d,j=10.8hz,2h),2.41(t,j=10.8hz,2h),1.88(td,j=12.6,4.2hz,2h),1.58(s,1h),1.53(s,1h)。
将式n所示化合物(295mg,0.99mmol)及氢氧化钯-碳(44.3mg,10%w/w)加入反应瓶中,随后加入20ml无水甲醇。用氢气球置换反应瓶中空气三次,随后在氢气球保护下于30℃搅拌6h。用硅藻土抽滤反应液,滤饼用甲醇洗涤,旋蒸滤液,蒸除溶剂,得白色固体(式vi-12所示化合物,168mg,收率:88.2%),无需纯化,可直接用于下一步;
(2)目标物(式ic-15所示化合物)的制备:
除4-氯丁酰氯替换实施例23中3-氯丙酰氯,及式vi-12所示化合物替换实施例23中式vi-1所示化合物外,其余步骤及试剂与实施例23相同,得到白色固体(式ic-15所示化合物的盐酸盐)85mg,收率:39.3%。
1hnmr(600mhz,dmso-d6,tms)δ10.68(s,1h),7.38(d,j=9.0hz,2h),7.00(d,j=7.2hz,1h),6.95(d,j=1.2hz,1h),6.93-6.89(m,3h),5.36(s,1h),3.74(s,3h),3.38-3.33(m,2h),3.21-3.12(m,2h),3.11-3.03(m,2h),2.71(dd,j=10.8,6.6hz,4h),2.54(d,j=7.8hz,2h),2.45-2.34(m,2h),1.81-1.70(m,6h),1.63-1.50(m,6h).
hrms(esi):m/z[m+h]+:408.2906。
实施例38
4-(2-萘基)1-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ic-16所示化合物)的制备:
除以2-溴萘替换实施例37中4-甲氧基溴苯外,其它步骤及试剂与实施例37相同,得到白色固体(式ic-16所示化合物的盐酸盐)102mg,收率:37.0%。
1hnmr(600mhz,dmso-d6,tms)δ10.86(s,1h),7.99(s,1h),7.94-7.88(m,3h),7.64(d,j=7.8hz,1h),7.54-7.47(m,2h),7.00(d,j=7.8hz,1h),6.96(d,j=1.2hz,1h),6.91(dd,j=7.8,1.2hz,1h),5.62(s,1h),3.39(s,2h),3.24(s,2h),3.10(s,2h),2.71(dd,j=10.8,7.8hz,4h),2.55(t,j=7.8hz,4h),1.87(d,j=13.8hz,2h),1.82-1.73(m,4h),1.64-1.57(m,2h),1.58-1.49(m,4h).
hrms(esi):m/z[m+h]+:428.2955。
实施例39
4-(1-萘基)1-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ic-17所示化合物)的制备:
除以1-溴萘替换实施例37中4-甲氧基溴苯外,其它步骤及试剂与实施例37相同,得到白色固体(式ic-17所示化合物的盐酸盐)120mg,收率:41.4%。
1hnmr(600mhz,dmso-d6,tms)δ10.35(s,1h),8.93-8.87(m,1h),7.96-7.90(m,1h),7.85(d,j=8.4hz,1h),7.56(d,j=7.2hz,1h),7.53-7.44(m,3h),7.00(d,j=7.2hz,1h),6.95(s,1h),6.90(d,j=7.2hz,1h),5.76(s,1h),3.49-3.33(m,2h),3.10(s,2h),2.79-2.64(m,4h),2.54(t,j=7.2hz,2h),2.24(d,j=13.2hz,2h),1.81-1.69(m,4h),1.63-1.57(m,2h),1.57-1.49(m,4h).
hrms(esi):m/z[m+h]+:428.2947。
实施例40
4-(4-吗啉苯基)-1-(4-(6,7,8,9-四氢-5h-苯并[7]轮烯-2-基)丁基)哌啶-4-醇(式ic-18所示化合物)的制备:
除以4-(4,-溴苯基)吗啉替换实施例37中4-甲氧基溴苯外,其它步骤及试剂与实施例37相同,得到白色固体(式ic-18所示化合物的盐酸盐)65mg,收率:30.3%。
1hnmr(600mhz,dmso-d6,tms)δ9.68(s,1h),7.30(d,j=8.4hz,2h),7.01(d,j=7.2hz,1h),6.96(s,1h),6.93(d,j=8.4hz,2h),6.91(d,j=7.2hz,1h),5.28(s,1h),3.75-3.72(m,4h),3.38(d,j=11.4hz,2h),3.23-3.15(m,2h),3.14-3.10(m,2h),3.09-3.06(m,4h),2.74-2.70(m,4h),2.55(t,j=7.8hz,2h),2.26-2.17(m,2h),1.82-1.75(m,4h),1.74-1.67(m,2h),1.63-1.51(m,6h).
hrms(esi):m/z[m+h]+:463.3318。
本发明化合物体外抗真菌活性测试
实施例41
(1)测试菌株:白色念珠菌(c.alb.sc5314),光滑念珠菌(c.gla.8535),新生隐球菌(c.neo.h99),及耐氟康唑白念珠菌(c.alb.0304103)。
(2)待测化合物:式ia-1~ia-14,式ib-1~ib-8及式ic-1~ic-18所示化合物;及对照化合物—氟康唑(flc)。
(3)材料及仪器:移液枪,96孔板,血细胞计数板,涡旋仪,玻璃管,ep管,超净化工作台(苏州安泰空气技术有限公司),thz-82a台式恒温振荡器(上海跃进医疗器械厂)。
(4)培养基配制:
yepd培养液:酵母浸膏10g,蛋白胨20g,葡萄糖20g,加三蒸水900ml溶解,加入2mg/ml氯霉素水溶液50ml,三蒸水定容至1000ml,高压灭菌(121℃,15min),后于4℃保存备用。
rpmi1640液体培养液:rpmi1640(gibcobrl)10g,nahco32.0g,吗啡啉丙磺酸(mops)34.5g,naoh2.7g,三蒸水定容至1000ml,抽滤灭菌,后于4℃保存备用。
pbs缓冲溶液:8.0gnacl,3.57gna2hpo4·12h2o,0.2gkcl,0.24gkh2po4,三蒸水定容至1000ml,高压灭菌(121℃,15min),室温保存备用。
(5)待测化合物的配制:待测化合物用dmso溶解,配制成2mg/ml的溶液,置于-20℃冰箱储存备用。
(6)菌悬液的制备:
用接种圈从4℃保存的sda培养基上挑取单克隆的菌株,接种至1mlyepd培养液中,于30℃,200rpm振荡培养,活化16h,使真菌处于指数生长期后期。取该菌液至1.5mlep管,离心(3000g,5min,4℃),弃上清液,用1ml的pbs缓冲液吹打混匀,用pbs缓冲溶液吹洗2次,用1mlrpmi1640培养液吹打混匀,用血细胞计数板计数,以rpmi1640培养液调整菌液浓度至1×103cfu/ml。
(7)检测:
将配制好的菌悬液加入96孔板中,第1列加入200μl,2至11列加入100μl。第12列加入1640培养基做空白对照。每个待测药物(2mg/ml)都取6.4μl于第一列菌悬液中,作三复孔,倍半稀释10个浓度梯度,即终浓度为64μg/ml-0.125μg/ml,第11列不加药,作阳性对照。
放入35℃恒温培养箱中孵育48h,用酶标仪测定od630处的吸光度。以阳性对照的od值为100%,最低抑菌浓度(mic50)为50%od值的最低化合物浓度。
协同指数fici测试方法参考clsi(以前称为nccls)(m27-a)的方法。本实施例中,用rpmi1640培养基稀释菌悬液至103cfu/ml,并将稀释好的菌悬液加入96孔板中。加入flc及目标化合物,倍半稀释使flc浓度从0.125μg/ml~64μg/ml,化合物浓度从2μg/ml~32μg/ml。菌悬液不加药组和空白培养基rpmi1640组分别作为阳性和阴性对照。将96孔板放入35℃恒温培养箱中孵育48h,用酶标仪测定od630处的吸光度,并算得mic50值。联合给药时flc的mic50除以单独给药时的mic50,加上联合给药时化合物的mic50除以单独给药时化合物的mic50即得协同指数fici。当fici≤0.5时,可以认为化合物和flc具有协同作用;当fici>4时,可以认为化合物和flc具有拮抗作用;当0.5<fici≤4时,可以认为化合物和flc具有既不发挥协同作用,也不发挥拮抗作用。
测试结果见表1(化合物体外抗真菌活性结果(mic50,μg/ml,48h)和协同指数(fici))。
表1
续表1
*fici为化合物与氟康唑联合用药的协同指数,fici<0.5两种药表现为协同作用;使用菌株为临床分离的耐氟康唑白念珠菌(c.alb.0304103)。
由表1可知:本发明部分化合物表现出良好的抗真菌活性特点,其中,化合物ic-2,ic-9,ic-10和ic-14对新生隐球菌抑制活性优于阳性对照物—氟康唑(flc),化合物ic-11和ic-12抑制活性与氟康唑相当。体外协同抗真菌活性实验结果显示,部分化合物与flc联用对临床分离的耐氟康唑白念珠菌(c.alb.0304103)均表现出良好的协同效果。
实施例42(本发明化合物被膜形成抑制实验)
(1)实验菌株:新生隐球菌(c.neo.h99);(2)待测化合物:化合物ic-9,及阳性对照物-氟康唑(flc);
(3)材料及仪器:移液枪,96孔板,血细胞计数板,涡旋仪,玻璃管,ep管,超净化工作台(苏州安泰空气技术有限公司),thz-82a台式恒温振荡器(上海跃进医疗器械厂)。
(4)培养基配制:参照实施例44,配制yepd培养液,rpmi1640液体培养液,及pbs缓冲溶液;
(5)待测化合物的配制:待测化合物用dmso溶解,配制成10mg/ml的溶液,置于-20℃冰箱储存备用;
(6)菌悬液的制备:参照实施例44,以rpmi1640培养液调配制菌液浓度为1×106cfu/ml的菌悬液。
(7)3,3'-[1-(苯氨酰基)-3,4-四氮唑]-二(4-甲氧基-6-硝基)苯磺酸钠)/甲萘醌(xtt/menadione)溶液的配制:
xtt用灭菌的pbs缓冲液配成0.5mg/ml的饱和溶液,用0.22um孔径的滤器过滤除菌,甲萘醌用100%丙酮配成10mm溶液,实验时向xtt溶液中加入10mm的甲萘醌使终浓度为1um,操作过程中要避光。
(8)测试步骤:
黏附:将菌液加入96孔板,1-11号孔,第12号不加,每孔100μl,37℃恒温箱静置培养90min。
另取一个96孔板,向其中加入空白1640培养液,第1列加入300ml,2至11列加入150ml。每个待测化合物(10mg/ml)都取1.28μl于第一列菌悬液中,作三复孔,倍半稀释10个浓度梯度,即终浓度为64μg/ml~0.125μg/ml,(第11列不加,作阳性对照)。
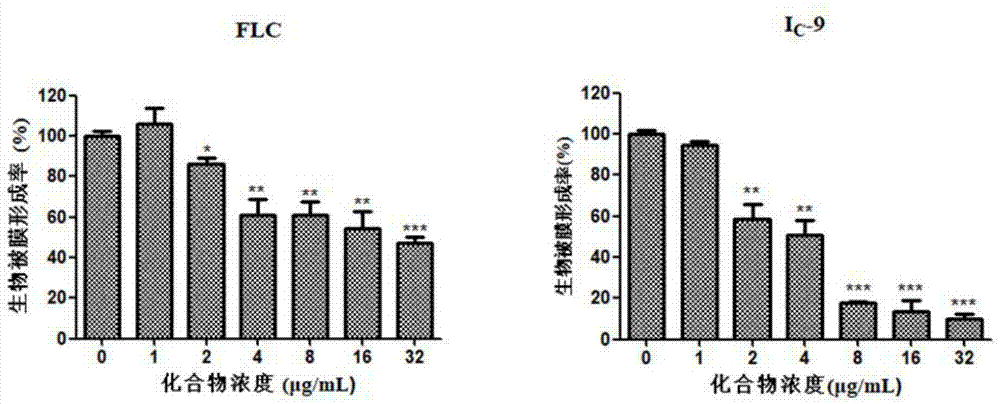
取出培养90min后的96孔板,弃上层rpmi1640培养液,用150μl的pbs缓冲液轻轻洗3次;取已配好的药物溶液100μl加入对应孔中(第11列只加100μl的rpmi1640做阳性对照,第12列不加)。37℃静置培养24h。测定黏附最低抑菌浓度(smic80):菌株生物被膜培养24h后,取出,用pbs缓冲液轻轻洗3次,每孔加入150μl的xtt/menadione溶液(第12列孔也加),避光,37℃孵育3h,取出,吸取70μl上清液至无菌96孔板,用酶标仪于490nm测各孔od值。与阳性孔比较,以od值下降80%以上的药物浓度最低孔中的药物浓度为smic80,测试结果如图1所示。
由图1可知,化合物ic-9对新生隐球菌(c.neo.h99)的被膜形成具有明显的抑制作用,且抑制效果优于阳性对照物—flc。
实施例43(本发明部分化合物的时间-生长曲线测试)
用接种圈从4℃保存的sda培养基上挑取单克隆的菌株(新生隐球菌(c.neo.h99)),接种至1mlyepd培养液中,于30℃,200rpm振荡培养,活化16h,使真菌处于指数生长期后期。取该菌液至1.5ml的ep管,离心(3000g,5min,4℃),弃上清液,用1ml的pbs缓冲液吹打混匀,用pbs缓冲溶液吹洗2次,用1ml的rpmi1640培养液吹打混匀,用血细胞计数板计数,以rpmi1640培养液调整菌液浓度至2×105cfu/ml。
取数只15ml离心管,加5ml菌浓度为2×105cfu/ml的1640培养基,加入梯度浓度的待测化合物ic-9和氟康唑(flc),空白对照加入相同体积的dmso,所有菌液中dmso含量均低于0.5%。30℃,200rpm振荡培养,分别于2、4、6、8、10、12、24和48h取10μl不同药物浓度的真菌悬液,以血细胞计数板计数。化合物ic-9和阳性对照物—氟康唑(flc)分别作用于新生隐球菌(c.neo.h99)的时间-生长曲线如图2所示。
由图2可知,化合物ic-9可以呈浓度依赖地抑制新生隐球菌的生长,且抑制作用优于阳性对照物—flc。
实施例44(本发明部分化合物小鼠系统性真菌感染实验)
选用雌性icr小鼠作为实验动物(约20g),经尾静脉注射给予1×106cfu/ml浓度的新生隐球菌(c.neo.h99)0.2ml(即2×105cfu/ml),造成系统性真菌感染模型。
一定浓度的待测物(化合物ic-9和阳性对照物—氟康唑(flc))经腹腔注射给药,1天1次给药,共给药5天,在第6天将小鼠处死、取脑、将脑组织匀浆均匀,将匀浆液稀释一定倍数后加入到sda培养基涂板,将培养基上的菌落计数,计算小鼠脑部真菌荷菌量。结果如表2和图3所示。
表2系统性新生隐球菌(c.neo.h99)感染小鼠模型实验结果
由表2及图3可知:化合物ic-9能够显著降低系统性真菌感染的小鼠的脑部荷菌量(p<0.001),降低隐球菌性脑膜炎的发生率。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种碱性离子液体催化制备吡喃并[4,3-b]吡喃衍生物的方法
- 全氟烷基苯并氮杂*并喹喔啉衍生物及其合成方法
- 苯并呋喃[2,3-b]吡嗪衍生物及其在有机电致荧光器件中的应用
- 一种手性2h-吡喃-2-酮衍生物的合成方法
- 用于促进脂肪细胞分化的包含羟基吡喃酮衍生物化合物的化妆品组合物的制作方法
- 吡喃并秋葵纤维异甲酚衍生物以及代谢综合征或炎症疾病治疗用药学组成物的制作方法
- 2-芳基-3-(4-羟基-2h-吡喃-2-酮-3-基)吲哚衍生物及其合成方法
- 一种基于苯并吡喃腈的可视化检测汞离子的探针的合成方法及应用
- 一种多取代苯并[4,5]咪唑并[1,2-b]吡唑衍生物的制备方法
- 一种含吡啶苯并硫氮杂卓衍生物、其制备方法及用图