一种制备聚烯烃共混物的方法与流程
本发明属于烯烃聚合反应技术领域,具体涉及一种制备超高分子量聚乙烯与聚烯烃共混物的方法。
背景技术:
聚烯烃共混物是将两种或两种以上的聚烯烃通过物理或化学的方法混合,进而达到提升材料综合性能的目的。如双峰聚乙烯、聚丙烯/聚乙烯、高密度聚乙烯/超高分子量聚乙烯、线性低密度聚乙烯/超高分子量聚乙烯等共混物。以两种聚乙烯的混合为例,高分子量聚乙烯具有优异的机械性能,然而加工性能较差;低分子量聚乙烯具有优异的加工性能,与高分子量聚乙烯共混时能够作为增塑剂,大幅改善共混物的加工性能。然而制约共混物性能优化的核心问题之一是两种高分子的界面作用,当两种高分子化学结构、支化结构、分子量差异越大时,这两种高分子的界面作用就越差。
链缠结是高分子凝聚态物理中十分重要的结构,很大程度上决定了高分子链段的松弛行为、链扩散、混合等行为,直接影响了高分子材料的结晶、热性能、机械性能和加工性能。以商用的超高分子量聚乙烯(uhmwpe)为例,商用的uhmwpe基底树脂多采用负载型齐格勒-纳塔(ziegler-natta)催化剂在淤浆聚合中进行。虽然催化剂活性高,但催化剂活性位随机分散于载体上,相距很近,增加了初生链段发生链交叠的概率。加之聚合温度≥60℃,致使初生的链段来不及结晶,极易形成链缠结。采用该法制备的1mg分子量为100万的商用uhmwpe初生颗粒中,缠结点含量可达1014个。大量的链缠结使得uhmwpe熔体粘度大,加工异常困难,限制了uhmwpe与其他聚烯烃之间的链扩散行为,制约了共混物界面作用强度的提升,共混物容易出现相分离,致使材料结构缺陷十分明显。
因此,降低聚烯烃初生粒子的链缠结程度,能够减少材料的结晶缺陷、降低熔体粘度,进而不仅可改善聚乙烯的加工性能,还能够降低材料分子链扩散壁垒。低缠结状态是亚稳态,共混物后续经熔体加工时,逐渐形成新的缠结点,并最终达到稳态缠结。在链缠结重新建立平衡的过程中,低缠结的组分有利于促进各组分间分子链的相互作用,形成更多的相间缠结、共结晶等,改善材料的界面作用。聚烯烃链缠结程度通常采用转子流变进行表征。链段缠结点间的平均分子量(me)与链缠绕密度成反比。me与橡胶平台区的弹性模量
其中gn为数量因子;ρ为密度;r为气体常数;t为绝对温度。温度一定的熔体中弹性模量的增加代表着链缠绕密度的增大。因此,样品初始平台模量与平台储能模量(储能模量为不随测试时间变化时的模量)之比
为此,申请公布号为cn105199214a的发明专利申请《一种聚乙烯共混材料的制备方法》公布了一种超高分子量聚乙烯与线性低密度聚乙烯共混材料的制备方法,包括先制备低缠结超高分子量聚乙烯,再将线性低密度聚乙烯、聚乙烯蜡及低缠结超高分子量聚乙烯加入转矩流变仪中共混均匀后挤出成型,得到的聚乙烯共混材料。与直接将商用的超高分子量聚乙烯和聚乙烯共混相比,该发明申请通过降低超高分子量聚乙烯的缠结点能提高共混物的均一性,大大改善了共混材料的机械性能及力学性能。然而,该方法中超高分子量聚乙烯的含量较少,使得共混材料的机械性能和力学性能受到一定的限制,且若提高超高分子量聚乙烯的加入量,可能会导致共混材料在后续加工中出现相分离。
技术实现要素:
本发明所要解决的技术问题是针对现有技术的现状,提供一种制备聚烯烃共混物的方法,以改善聚烯烃间的共混效果,进而提升共混物的机械性能和加工性能。
本发明解决上述技术问题所采用的技术方案为:一种制备聚烯烃共混物的方法,其特征在于包括如下步骤:
1)制备重均分子量为1200~350000g/mol的聚烯烃a,所述聚烯烃a在160℃下的起始储能模量与平台储能模量之比为0.1~0.95;
2)制备重均分子量为200000~10000000g/mol的聚烯烃b,所述聚烯烃b在160℃下的起始储能模量与平台储能模量之比为0.1~0.8;
3)通过化学反应将上述聚烯烃a与聚烯烃b共混,得到聚烯烃共混物c,所述聚烯烃共混物c在160℃下的起始储能模量与平台储能模量之比为0.1~0.75;且聚烯烃共混物c中聚烯烃b的含量为5~50wt%;
所述化学反应为聚合反应。
在上述方案中,所述聚烯烃b的重均分子量优选为500000~6000000g/mol。
作为优选,所述化学反应为聚合反应,制备聚烯烃共混物c的方法具体包括如下步骤:
(1)提供两个聚合反应器,分别为第一反应器和第二反应器,第一反应器为高压管示反应器、流化床反应器、搅拌反应釜中的一种,第二反应器为高压管示反应器、流化床反应器、搅拌反应釜中的一种;
(2)将烯烃、氢气和催化剂a通入第一反应器中进行反应,制得聚烯烃a,其中氢气与烯烃的摩尔比为0.01~0.5;
(3)将步骤(2)制得的聚烯烃a传送至第二反应器,并将烯烃通入第二反应器中进行反应,制得聚烯烃b,进而获得聚烯烃b改性的聚烯烃a,即聚烯烃共混物c;
其中,所述步骤(2)、(3)的反应温度分别为0~100℃,反应压力分别为1~60bar,反应时间分别为1min~8h;所述烯烃选自乙烯或α-烯烃中的至少一种;所述催化剂a为茂金属催化剂、齐格勒-纳塔催化剂、过渡金属催化剂、无机铬催化剂、有机铬催化剂中的一种。
较优选的是,所述步骤(3)中在第二反应器内加入催化剂b,该催化剂b为茂金属催化剂、齐格勒-纳塔催化剂、过渡金属催化剂、无机铬催化剂、有机铬催化剂中的一种。
同样优选的是,所述化学反应为聚合反应,制备聚烯烃共混物c的方法具体包括如下步骤:
(1)提供一个聚合反应器,该聚合反应器选自流化床反应器或搅拌反应釜;
(2)将烯烃、催化剂b通入聚合反应器进行反应,制得聚烯烃b;
(3)将步骤(2)反应制得的气体从聚合反应器内分离;
(4)将烯烃、氢气、催化剂a通入聚合反应器内进行反应,其中氢气与烯烃的摩尔比为0.01~0.5,制得聚烯烃a,进而获得聚烯烃共混物c。
上述步骤(2)和(4)的反应温度分别为0~100℃,反应压力分别为1~60bar,反应时间分别为1min~8h;所述烯烃选自乙烯或α-烯烃中的至少一种;所述催化剂a、b分别为茂金属催化剂、齐格勒-纳塔催化剂、过渡金属催化剂、无机铬催化剂、有机铬催化剂中的一种。
改进,所述催化剂a和催化剂b均为齐格勒-纳塔催化剂,所述齐格勒-纳塔催化剂的制备包括如下步骤:
(一)将多孔载体与醇在四氢呋喃溶液中混合4~8h后,得到物质a,其中所述醇为乙醇、丙醇、1-丁醇、乙二醇、丙二醇、1,4丁二醇中的至少一种;
(二)将纳米粒子与mg化合物在四氢呋喃溶液中混合1~4h后,加入上述物质a,其中mg化合物为氯化镁、溴化镁、乙氧基镁、氢氧化镁中的一种或二种,醇与mg原子的摩尔比为1~10,所述纳米粒子与上述多孔载体的重量比为1~90wt%,上述多孔载体与mg化合物的重量比为0.1~10,搅拌1~8h,过滤,将固体粉末干燥至自由流动,得到载体a;
(三)将过量的烷基铝加入至步骤(二)中所得的载体a,搅拌4~8h,用四氢呋喃溶液洗涤3~8次,除去多余的烷基铝,将固体粉末干燥至自由流动,得到载体b;
(四)将ticl4或ticl3的四氢呋喃溶液加入至步骤(三)中所得的载体b,其中ti原子与载体b的重量比为0.1~10wt%,搅拌4~8h后,用四氢呋喃溶液洗涤固体粉末1~10次,干燥至自由流动,即获得齐格勒-纳塔催化剂。
再改进,所述步骤(一)中的多孔载体选自二氧化硅、氧化铝、氧化锆、二氧化钛、二氧化硅-氧化铝、蒙拓土中的至少一种;所述步骤(二)中的纳米粒子为纳米碳管、二氧化硅、二氧化钛、氧化钙、石墨烯、fe3o4、c3n4和聚倍半硅氧烷中的至少一种;所述步骤(三)中的烷基铝包括三甲基铝、三甲基铝氧烷、三乙基铝中的一种或二种。上述醇与mg原子的摩尔比为1~10;al原子与ti原子摩尔比为10~200。
进一步改进,所述纳米粒子表面经亲核基团改性,该亲核基团包括羟基、氨基、羧基、酯基中的至少一种;所述纳米粒子中亲核基团的含量为0.01~10wt%,优选0.01~1wt%;所述纳米粒子的尺寸为1~100nm。
当催化剂a和/或b选用上述齐格勒-纳塔催化剂时,优选的是,在反应时同时添加助催化剂,所述助催化剂包括三乙基铝、三异丁基铝、1氯二乙基铝、三甲基铝氧烷中的至少一种,优选为三乙基铝。
与现有技术相比,本发明的优点在于:本发明将两种聚烯烃通过聚合反应共混混合,与现有机械共混法相比,该法能够改善两种聚烯烃的混合尺度,实现分子链尺度的均匀混合;且通过保证聚烯烃各组分的低缠结特性,最终制备缠结程度低的共混物,低缠结的共混物在加工过程中有利于促进共混组分间链扩散行为,强化共混组分形成新的链缠绕结构,有利于促进共混物的界面相互作用,能够大幅提升共混物的机械性能和加工性能;本发明通过化学反应共混来制备低缠结的聚烯烃共混物,使得共混物不存在相分离,且随着(超)高分子量聚烯烃加入量的增加,共混物仍具有均一的共混效果。
附图说明
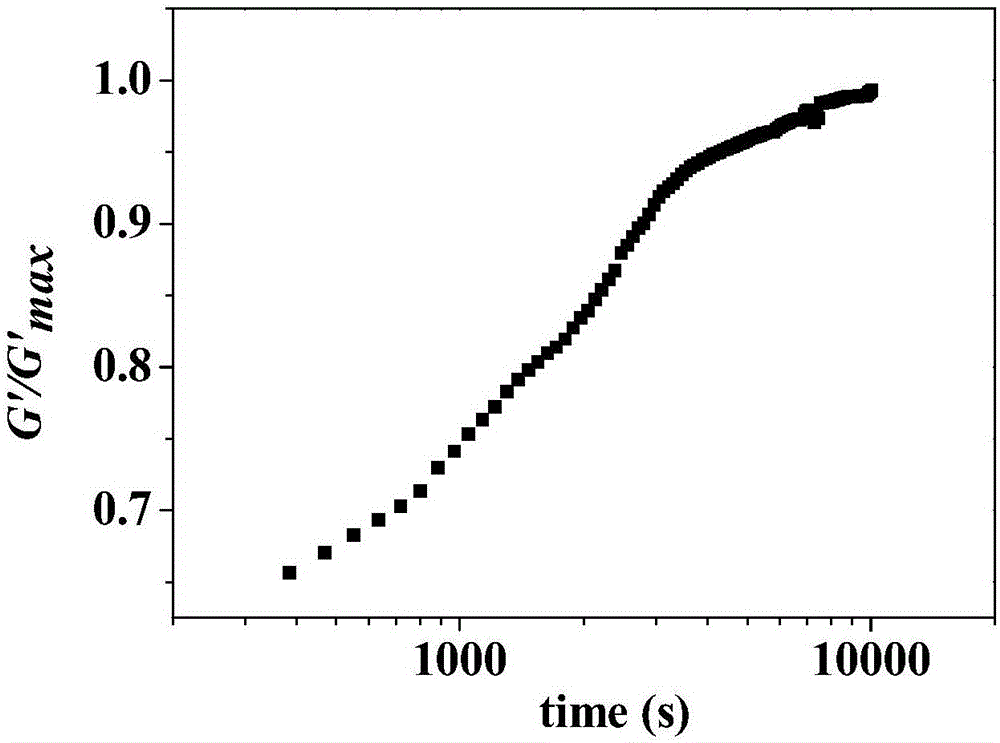
图1为本发明实施例1中第一反应器内制备的聚烯烃a的储能模量随时间的建立曲线;
图2为本发明对比例1、对比例2与实施例1中所得聚烯烃共混物的储能模量(g’)与损耗模量(g”)间的关系图。
具体实施方式
以下结合附图实施例对本发明作进一步详细描述。
实施例1:
一种制备聚烯烃共混物的方法,其特征在于包括如下步骤:
(1)将1.0g的多孔载体sio2与6mmol的1,4-丁二醇在20ml四氢呋喃thf溶液中混合4h后,再用20ml的thf溶液洗涤5次,获得物质a(即1,4-丁二醇吸附的多孔载体);
(2)将质量为0.5g平均粒径为10nm的纳米二氧化钛(表面羟基含量0.01wt%);与6mmol的mgcl2(1,4-丁二醇与镁原子的摩尔比1)混合,在20mlthf中混合4h,加入上述物质a,在thf中搅拌8h后,过滤,将固体粉末干燥至自由流动,得到载体a;
(3)将10mmol三乙基铝加入至步骤(2)中所得的载体a,搅拌8h,用thf洗涤8次,除去多余的三乙基铝,将固体粉末干燥至自由流动得到载体b;
(4)将ticl4的thf溶液加入至步骤(3)中所得载体b,其中ti原子与载体b的重量比为6wt%,搅拌4h后,用thf洗涤固体粉末10次,干燥至自由流动,获得催化剂;
(5)提供两个聚合反应器,分别为第一反应器(高压管示反应器)和第二反应器(流化床反应器),将第一反应器加热至80℃,调节聚合压力为1bar,依次加入200ml甲苯、5ml三乙基铝(al原子与ti原子的摩尔比100)、催化剂、乙烯、1-丁烯和氢气,其中1-丁烯与乙烯摩尔比1,氢气与烯烃(乙烯、1-丁烯)的摩尔比为0.1。反应30min后,得到聚烯烃a,所得聚烯烃a重均分子量为200000g/mol,且在160℃下经转子流变仪测定模量随时间变化曲线,具体请参见图1,可得起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
(6)将反应产物聚烯烃a转移至第二反应器中,且将第二反应器的温度调至60℃,反应压力调至10bar,并加入催化剂、乙烯、3ml三乙基铝,反应2h后,继续制备聚烯烃b(即超高分子量聚乙烯uhmwpe),获得uhmwpe改性的聚烯烃a,即聚烯烃共混物c。第二反应器制备的聚烯烃b,分子量达6000000g/mol,
实施例2:
一种制备聚烯烃共混物的方法,其特征在于包括如下步骤:
(1)将1.0g的多孔载体tio2与6mmol的乙二醇在20ml四氢呋喃thf溶液中混合6h后,再用20ml的thf溶液洗涤醇改性的多孔载体5次,获得物质a(即乙二醇吸附的多孔载体);
(2)将质量为0.9g平均粒径为100nm的纳米碳管(表面羟基含量5wt%);与6mmol的溴化镁(乙二醇与镁原子的摩尔比1)混合,在20mlthf中混合2h,加入上述物质a,在thf中搅拌6h后,过滤,将固体粉末干燥至自由流动,得到载体a;
(3)将10mmol三甲基铝加入至步骤(2)中所得的载体a,搅拌5h,用thf洗涤5次,除去多余的三甲基铝,将固体粉末干燥至自由流动得到载体b;
(4)将ticl4的thf溶液加入至步骤(3)中所得载体b,其中ti原子与载体b的重量比为10wt%,搅拌5h后,用thf洗涤固体粉末5次,干燥至自由流动,获得催化剂;
(5)提供两个聚合反应器,分别为第一反应器(流化床反应器)和第二反应器(搅拌反应釜),将第一反应器加热至80℃,调节聚合压力为60bar,加入催化剂、乙烯、1-丁烯和氢气,其中1-丁烯与乙烯摩尔比0.01,氢气与烯烃(乙烯、1-丁烯)的摩尔比为0.5。反应8h后,得到聚烯烃a,所得聚烯烃a重均分子量为1200g/mol,且在160℃下经转子流变仪测定模量随时间变化曲线,可得起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
(6)将反应产物聚烯烃a转移至第二反应器中,且将第二反应器的温度调至0℃,反应压力调至1bar,并将乙烯经第二反应器的进料口通入第二反应器中进行反应,反应1min,制备聚烯烃b,进而获得聚烯烃共混物c,第二反应器制备的聚烯烃b分子量达10000000g/mol,
实施例3:
一种制备聚烯烃共混物的方法,其特征在于包括如下步骤:
(1)将1.0g的多孔载体氧化铝与5mmol的乙醇在20ml四氢呋喃thf溶液中混合8h后,再用20ml的thf溶液洗涤醇改性的多孔载体5次,获得物质a(即乙醇吸附的多孔载体);
(2)将质量为0.01g平均粒径为1nm的纳米二氧化硅(表面氨基含量10wt%)与0.5mmol的溴化镁(乙醇与镁原子的摩尔比10)混合,在20mlthf中混合1h,加入上述物质a,在thf中搅拌1h后,过滤,将固体粉末干燥至自由流动,得到载体a;
(3)将10mmol三甲基铝氧烷加入至步骤(2)中所得的载体a,搅拌4h,用thf洗涤3次,除去多余的三乙基铝,将固体粉末干燥至自由流动得到载体b;
(4)将ticl3的thf溶液加入至步骤(3)中所得载体b,其中ti原子与载体b的重量比为0.1wt%,搅拌8h后,用thf洗涤固体粉末1次,干燥至自由流动,获得催化剂;
(5)提供一个聚合反应器(流化床反应器),将聚合反应器加热至100℃,调节聚合压力为20bar,依次加入催化剂和乙烯,反应1min后,得到聚烯烃b,所得聚烯烃b重均分子量为200000g/mol,且在160℃下经转子流变仪测定模量随时间变化曲线,可得起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
(6)将步骤(5)反应后的气体从反应器内分离,再将乙烯、氢气、催化剂通入聚合反应器内进行反应,其中氢气与乙烯的摩尔比为0.01,反应温度为0℃,压力为1bar,,时间为8h,制得聚烯烃a,进而获得聚烯烃共混物c,所得聚烯烃a重均分子量为350000g/mol,且在160℃下经转子流变仪测定模量随时间变化曲线,可得起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
实施例4:
一种制备聚烯烃共混物的方法,其特征在于包括如下步骤:
(1)将1.0g的多孔载体氧化锆与8mmol的1-丁醇在20ml四氢呋喃thf溶液中混合6h后,再用20ml的thf溶液洗涤醇改性的多孔载体5次,获得物质a(即1-丁醇吸附的多孔载体);
(2)将质量为0.7g平均粒径为50nm的石墨烯(表面羟基含量5wt%)与7mmol的乙氧基镁(1-丁醇与镁原子的摩尔比1)混合,在20mlthf中混合2h,加入上述物质a,在thf中搅拌6h后,过滤,将固体粉末干燥至自由流动,得到载体a;
(3)将5mmol三乙基铝、5mmol三甲基铝加入至步骤(2)中所得的载体a,搅拌5h,用thf洗涤5次,除去多余的三乙基铝、三甲基铝,将固体粉末干燥至自由流动得到载体b;
(4)将ticl4的thf溶液加入至步骤(3)中所得载体b,其中ti原子与载体b的重量比为10wt%,搅拌5h后,用thf洗涤固体粉末5次,干燥至自由流动,获得催化剂;
(5)提供两个聚合反应器,分别为第一反应器(流化床反应器)和第二反应器(搅拌反应釜),将第一反应器的温度调至0℃,调节聚合压力为30bar,加入催化剂、乙烯、1-丁烯和氢气,其中1-丁烯与乙烯摩尔比0.5,氢气与烯烃(乙烯、1-丁烯)的摩尔比为0.2。反应10min后,得到聚烯烃a,所得聚烯烃a重均分子量为12000g/mol,且在160℃下经转子流变仪测定模量随时间变化曲线,可得起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
(6)将反应产物聚烯烃a转移至第二反应器中,且将第二反应器加热至100℃,反应压力调至10bar,并将乙烯经第二反应器的进料口通入第二反应器中进行反应,反应8h,制备聚烯烃b,进而获得聚烯烃共混物c,第二反应器制备的聚烯烃b分子量达500000g/mol,
实施例5:
与实施例2基本相同,区别在于本实施例的催化剂直接选用茂金属催化剂,该茂金属催化剂以过渡金属ti元素配合物作为主催化剂,以有机硼化物b(c6f5)3作为助催化剂。由本实施例制得的聚烯烃共混物c起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
实施例6:
与实施例2基本相同,区别在于本实施例的催化剂直接选用过渡金属催化剂ni(co)4。由本实施例制得的聚烯烃共混物c起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
实施例7:
与实施例2基本相同,区别在于本实施例的催化剂直接选用无机铬催化剂,即phillips型氧化铬催化剂。由本实施例制得的聚烯烃共混物c起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
实施例8:
与实施例2基本相同,区别在于本实施例的催化剂直接选用有机铬催化剂:双三苯基硅烷铬酸酯。由本实施例制得的聚烯烃共混物c起始储能模量与平台储能模量(储能模量不随测试时间变化时的模量)之比
对比例1:
将分子量为200000g/mol的聚乙烯与分子量为2300000g/mol的商业uhmwpe按质量比1:1混合后,经双螺杆挤出机,在温度230℃、转速160转/min,混合20min后,得到uhmwpe改性的聚烯烃。经测试,聚烯烃共混物的拉伸强度为40.5mpa,断裂伸长率为3.5%。
对比例2:
将分子量为200000g/mol的聚乙烯与低缠结超高分子量聚乙烯(分子量为2000000g/mol,
如图2所示,对比例1、2与实施例1中所得的聚烯烃共混物中超高分子量聚乙烯占共混物的50%。然而,对比例1和2中,g’~g”为曲线,表明共混物中存在明显的相分离;实施例1中,g’~g”为直线,随着g”的增加,g’线性增加,表明,即使在如此大的uhmwpe的混入量时(50wt%),共混物仍能具有均一的共混效果,不存在相分离。现有制备聚烯烃共混物的方法中,uhmwpe的最高加入量为10wt%,超过这个量,共混物就会存在如对比例1、2中的相分离。进一步说明本发明制得的聚烯烃共混物中uhmwpe的含量明显提高,且不存在相分离。
本发明上述各实施例和对比例在对产品的加工性能及力学性能进行测试时,满足以下条件:
所有空气敏感物质的操作均采用标准真空双排线无水无氧操作方法;所用试剂均需精制处理后使用。
聚合物的拉伸模量和断裂伸长率根据国标gb/t1040测得。
聚合物的重均分子量根据凝胶渗透色谱polymerlaboratoriespl-220仪器测定。管柱及旋转格室在140℃操作。溶剂是1,2,4-三氯苯,聚合物浓度为3‰。注射体积100微升,流速1.0毫升/分钟。
聚合物的低缠结特性由流变学测试。流变学测试由一个轴应变流变仪(haakeiiiinstrument)测定。具体测定方法为:在120℃、20tons条件下将聚乙烯粉料压片30min,制得直径8mm样品,用于流变学研究。流变仪平行板间的底盘在氮气环境下加热到160℃,稳定5min开始流变实验。动态频率扫描以固定频率1hz测试。动态时间扫描以固定1rad/s测试。根据旋转流变在160℃下测定的储能模量随时间的建立曲线中,起始储能模量与最大储能模量(储能模量不在随时间变化,体系达到热力学稳定状态)之比
- 还没有人留言评论。精彩留言会获得点赞!