一种由铜化合物诱导制备菲啶酮类化合物的方法与流程
本发明属于有机合成、金属催化领域,特别涉及一种由铜化合物诱导制备菲啶酮类化合物的方法。
背景技术:
菲啶酮是一种作为异喹啉衍生物的基本母核结构,该类化合物及其衍生物具有特殊的生物活性。其中,苯并菲啶酮类化合物所具有的显著的抗癌活性,在医药领域具有广泛的用途,从而引起了医药研究人员的极大兴趣。因此,开发菲啶酮的合成方法具有重要的实用价值。
2006年,furuta等(organicletters,2006,9(2),183-186)报道了以两分子邻溴苯甲酰胺在cs2co3、pd(oac)2和配体的共同作用下反应得到了一系列的菲啶酮衍生物。反应式如式1所示。此反应的催化剂和原料价格相对昂贵,配体选择直接影响产率的高低,生产成本很高,不利于大规模的制备。
2010年,王官武等(angewandtechemie,2011,50(6),1380-1383)报道了用碘苯和苯甲酰胺衍生物在三氟乙酸、醋酸钯和氧化银的共同作用下合成了一系列菲啶酮衍生物。反应式如式2所示。该反应高效一步合成菲啶酮,不过该反应要用到三氟乙酸作溶剂要求苛刻且高温反应下对设备要求较高,难以实现工业化生产。
2012bhakuni等(organicletters,2012,14(11),2838-2841)用n-苯基邻碘苯甲酰胺在偶氮二异腈,叔丁醇钾作用下催化分子内成环构建菲啶酮,反应式如式3所示。但是此合成方法需要在较高的温度下进行,并且苯对人体的危害较大,限制了其在有机合成中的应用。
2014年,yang等(organic&biomolecularchemistry,2014,48(10),5351-5355)使用邻碘苯甲酰胺衍生物与2-(三甲基硅)苯基三氟甲烷磺酸盐在醋酸钯、三(邻甲基)苯基磷为配体、氟化铯作用下实现菲啶酮环的构建,反应式如式4所示。但是此合成方法需要的邻位碘代物原料以及所使用的钯试剂价格昂贵,限制了其在有机合成和化学工业中的应用。
2017年,banerji等(europeanjournalorganicchemistry,2017,48(35),5214-5218)用苯甲酰胺衍生物和邻碘溴苯在碳酸铯、双三苯基磷二氯化钯的共同作用下合成了菲啶酮,反应式如式5所示。但是此合成方法需要的原料邻碘溴苯和钯试剂价格昂贵,不利于工业化生产。
现有技术中合成菲啶酮类化合物的反应条件较为苛刻,反应原料大多数为邻卤代的化合物,价格昂贵且不易得。因此使得它们在工业生产中受到了一定的限制。
技术实现要素:
为了克服上述现有技术的缺点与不足,本发明的首要目的在于提供一种由铜化合物诱导制备菲啶酮类化合物的方法。该方法是在氮气和密闭环境中,在铜盐的存在下,苯甲酰胺衍生物在铜化合物的诱导作用下与邻溴苯硼酸类化合物发生c-h活化串联反应生成菲啶酮类化合物;该方法使用“一锅煮”方法完成,原料也简单易得,反应操作简单,产率高,有利于工业化生产。
本发明的目的通过下述方案实现:
一种由铜化合物诱导制备菲啶酮类化合物的方法,包括以下步骤:将底物苯甲酰胺衍生物和邻溴苯硼酸类化合物、铜盐、碱、溶剂在惰性气氛下加入到反应容器中,然后加热反应,反应结束后将所得反应液纯化即得所需的菲啶酮类化合物。
所述的苯甲酰胺衍生物具有式6所示结构:
其中,r1表示其所连接的苯环上1个或多个取代基,各个r1彼此独立地选自氢、c1-c6烷基、c1-c6烷氧基、c1-c6酰基、c1-c6酯基、卤素、氰基、三氟甲基、c3-c6环烷基、c5-c14芳基、c5-c14杂芳基。
所述的邻溴苯硼酸类化合物具有式7所示结构:
其中,r2表示其所连接的苯环上1个或多个取代基,r2彼此独立地选自氢、c1-c6烷基、c1-c6烷氧基、c1-c6酰基、c1-c6酯基、卤素、三氟甲基。
所述的铜盐为cu(oac)2、cucl2、cubr2、cu2(oh)2co3、cuso4·5h2o或cu(oac)2·h2o中的至少一种,优选为cu(oac)2。
所述的碱为na2co3、k2co3、cs2co3、li2co3、naoac、nahco3、k3po4、koac中的至少一种,优选为cs2co3。
所述的溶剂为二甲基亚砜、n-甲基吡咯烷酮、甲醇或乙醇中的至少一种,优选为二甲基亚砜。
所述的惰性气氛为氮气、氩气、氖气中的至少一种,优选为氮气。
所述的苯甲酰胺衍生物和邻溴苯硼酸类化合物的用量满足:邻溴苯硼酸类化合物的摩尔用量为苯甲酰胺衍生物摩尔用量的1~3倍,优选为1.5倍;
所述的铜盐的用量满足:铜盐的摩尔用量为苯甲酰胺衍生物摩尔用量的1~3倍,优选为2.0倍;
所述的碱的用量满足:碱的摩尔用量为苯甲酰胺衍生物摩尔用量的1~4倍,优选为2倍;
所述的溶剂的用量满足:每1mmol的苯甲酰胺衍生物对应使用2~5ml的溶剂,优选为每1mmol的苯甲酰胺衍生物对应使用3ml的溶剂。
所述的加热反应是指加热至50~120℃下反应1~8h,优选为加热至100℃反应4h。
所述的纯化是指将所得反应液冷却至室温,加入乙酸乙酯稀释后过闪柱除去金属盐,将所得的有机层用水洗,然后用无水硫酸钠干燥、过滤,将所得滤液减压蒸馏后经硅胶柱层析分离,即得纯化后的产物。
所述的闪柱所用洗脱剂为乙酸乙酯;所述的柱层析所用的洗脱剂为体积比为1:8的乙酸乙酯和石油醚的混合溶液。
本发明的制备方法的反应式如式8所示:
即本发明所述的菲啶酮类化合物的结构式为:
本发明相对于现有技术,具有如下的优点及有益效果:
本发明需在氮气的环境下,用简单的铜化合物诱导苯甲酰胺衍生物与邻溴苯硼酸类化合物进行c-h活化串联反应就可得到菲啶酮类化合物,该方法所使用的原料、铜盐和碱简单易得,并且反应操作简单,产率高。通过本发明可以在温和条件下高产率得到一系列的菲啶酮类化合物,其中铜诱导的c-h键活化串联反应在天然产物、药物及农药等的合成中都具有广阔的应用前景,且有利于工业化生产发展。
附图说明
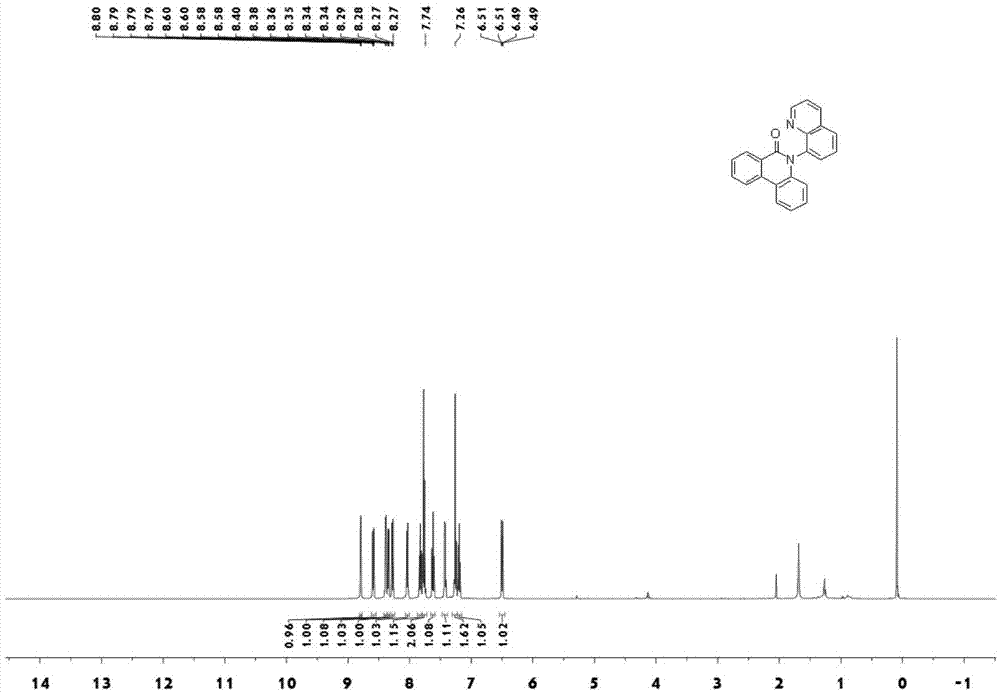
图1为实施例1中制备的5-(喹啉-8-基)菲啶-6(5h)-酮的核磁氢谱图;
图2为实施例1中制备的5-(喹啉-8-基)菲啶-6(5h)-酮的核磁碳谱图;
图3为实施例1中制备5-(喹啉-8-基)菲啶-6(5h)-酮的反应路线图;
图4为实施例2中制备的9-甲基-5-(喹啉-8-基)菲啶-6(5h)-酮的核磁氢谱图;
图5为实施例2中制备的9-甲基-5-(喹啉-8-基)菲啶-6(5h)-酮的核磁碳谱图;
图6为实施例3中制备的9-甲氧基-5-(喹啉-8-基)菲啶-6(5h)-酮的核磁氢谱图;
图7为实施例3中制备的9-甲氧基-5-(喹啉-8-基)菲啶-6(5h)-酮的核磁碳谱图;
图8为实施例4中制备的9-氟-5-(喹啉-8-基)菲啶-6(5h)-酮的核磁氢谱图;
图9为实施例4中制备的9-氟-5-(喹啉-8-基)菲啶-6(5h)-酮的核磁碳谱图;
图10为实施例5中制备的9-氯-5-(喹啉-8-基)菲啶-6(5h)-酮的核磁氢谱图;
图11为实施例5中制备的9-氯-5-(喹啉-8-基)菲啶-6(5h)-酮的核磁碳谱图;
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例中所用试剂如无特殊说明均可从市场常规购得。
实施例1:5-(喹啉-8-基)菲啶-6(5h)-酮
将248mg(1mmol)n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)促进剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:3做洗脱剂)得255mg黄色固体,产率为79%。
所得产物的各项表征数据如下:
m.p.=227~228℃;
核磁氢谱图如附图1所示,具体数据为1hnmr(500mhz,cdcl3)δ8.79(dd,j=4.2,1.7hz,1h),8.59(dd,j=8.0,1.0hz,1h),8.39(d,j=8.2hz,1h),8.35(dd,j=8.0,1.4hz,1h),8.28(dd,j=8.3,1.7hz,1h),8.07-8.00(m,1h),7.83(ddd,j=8.4,7.2,1.4hz,1h),7.80-7.72(m,2h),7.66-7.59(m,1h),7.42(dd,j=8.3,4.2hz,1h),7.31-7.22(m,2h),7.20(ddd,j=8.5,7.2,1.5hz,1h),6.50(dd,j=8.3,1.0hz,1h);
核磁碳谱图如附图2所示,具体数据为13cnmr(126mhz,cdcl3):δ161.97,151.38,144.52,139.47,136.30,136.06,134.38,132.73,130.52,129.75,129.38,129.07,129.00,127.92,126.75,125.95,123.03,122.44,121.90,121.84,119.07,116.86;
hrms(esi)calcdforc22h14n2o:[m+h]+322.1106,found:322.1140.
上述数据说明本实施例成功合成了5-(喹啉-8-基)菲啶-6(5h)-酮。
原料n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.4ml苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到4.1克n-(喹啉-8-基)苯甲酰胺,产率为80%。
原料n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=82~83℃;
1hnmr(500mhz,cdcl3):δ10.73(s,1h),8.95-8.93(m,1h),8.84-8.82(m,1h),8.17-8.15(m,1h),8.09-8.07(m,2h),7.60-7.51(m,5h),7.46-7.43(m,1h);
13cnmr(125mhz,cdcl3):δ165.41,148.22,138.80,136.38,135.24,134.61,131.81,128.87,128.02,127.45,127.36,121.64,116.58;
hrms(esi)calcdforc16h13n2o:[m+h]+249.1028,found:249.1030.
上述数据说明成功合成了原料n-(喹啉-8-基)苯甲酰胺。
以实施例1为例,本发明的反应路线图如图3所示。
实施例2:9-甲基-5-(喹啉-8-基)菲啶-6(5h)-酮
将262mg(1mmol)4-甲基-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得245mg黄色固体,产率为76%。
所得产物的各项表征数据如下:
m.p.=261~262℃;
核磁氢谱图如附图4所示,具体数据为1hnmr(500mhz,cdcl3)δ8.79(dd,j=4.2,1.7hz,1h),8.47(d,j=8.1hz,1h),8.33(dd,j=8.0,1.3hz,1h),8.27(dd,j=8.3,1.7hz,1h),8.17(s,1h),8.03(dd,j=7.6,2.1hz,1h),7.79-7.71(m,2h),7.43(td,j=8.1,2.5hz,2h),7.28-7.21(m,1h),7.18(ddd,j=8.5,7.2,1.5hz,1h),6.48(dd,j=8.3,1.0hz,1h),2.60(s,3h);
核磁碳谱图如附图5所示,具体数据为13cnmr(126mhz,cdcl3)δ162.02,151.33,144.54,143.23,139.58,136.30,136.11,134.35,130.57,129.72,129.34,129.31,129.03,128.84,126.73,123.65,122.96,122.31,121.89,121.86,119.05,116.81,22.19;
hrms(esi)calcdforc23h16n2o:[m+h]+336.1263,found:336.3859.
上述数据说明本实施例成功合成了9-甲基-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料4-甲基-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.8ml4-甲基苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到4.3克4-甲基-n-(喹啉-8-基)苯甲酰胺,产率为82%。
原料4-甲基-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=119~120℃;
1hnmr(500mhz,cdcl3):δ10.71(s,1h),8.94-8.92(m,1h),8.84-8.82(m,1h),8.16-8.14(m,1h),7.99-7.97(d,j=8.0hz,2h),7.52-7.50(m,1h),7.46-7.45(m,1h),7.44-7.43(m,1h),7.34-7.32(d,j=8.0hz,2h),2.44(s,3h);
13cnmr(125mhz,cdcl3):δ165.43,148.21,142.36,138.79,136.35,134.78,132.32,129.40,127.92,127.42,127.38,121.66,121.50,116.45,21.57;
hrms(esi)calcdforc17h15n2o:[m+h]+263.1184,found:263.1185.
上述数据说明成功合成了原料4-甲基-n-(喹啉-8-基)苯甲酰胺。
实施例3:9-甲氧基-5-(喹啉-8-基)菲啶-6(5h)-酮
将278mg(1mmol)4-甲氧基-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得175mg黄色固体,产率为52%。
所得产物的各项表征数据如下:
m.p.=266~268℃;
核磁氢谱图如附图6所示,具体数据为1hnmr(500mhz,cdcl3)δ8.80(dd,j=4.2,1.7hz,1h),8.51(d,j=8.8hz,1h),8.27(ddd,j=7.9,3.9,1.5hz,2h),8.03(dd,j=7.4,2.3hz,1h),7.82-7.68(m,3h),7.42(dd,j=8.3,4.2hz,1h),7.25-7.13(m,3h),6.47(dd,j=8.3,1.0hz,1h),4.02(s,3h);
核磁碳谱图如附图7所示,具体数据为13cnmr(126mhz,cdcl3)δ163.25,161.78,151.39,144.66,139.93,136.37,136.31,136.17,131.30,130.66,129.76,129.32,129.16,126.76,123.07,122.23,121.88,119.69,118.93,116.93,115.76,104.78,55.58;
hrms(esi)calcdforc23h16n2o2:[m+h]+352.1212,found:352.3853.
上述数据说明本实施例成功合成了9-甲氧基-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料4-甲氧基-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.9ml4-甲氧基苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到3.6克4-甲氧基-n-(喹啉-8-基)苯甲酰胺,产率为65%。
原料4-甲氧基-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=119~120℃;
1hnmr(500mhz,cdcl3):δ10.68(brs,1h),8.93(dd,j=7.5,1.5hz,1h),8.85(dd,j=4.2,1.7hz,1h),8.18(dd,j=8.3,1.7hz,1h),8.06-8.04(m,2h),7.62-7.45(m,3h),7.07-7.02(m,2h),3.90(s,3h);
13cnmr(125mhz,cdcl3):δ164.83,162.46,148.14,138.66,136.26,134.70,129.09,127.91,127.36,127.33,121.58,121.35,116.23,113.93,55.39;
hrms(esi)calcdforc17h15n2o2:[m+h]+279.1134,found:279.1132。
上述数据说明成功合成了原料4-甲氧基-n-(喹啉-8-基)苯甲酰胺。
实施例4:9-氟-5-(喹啉-8-基)菲啶-6(5h)-酮
将266mg(1mmol)4-氟-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得262mg黄色固体,产率为77%。
所得产物的各项表征数据如下:
m.p.=245~246℃;
核磁氢谱图如附图8所示,具体数据为1hnmr(500mhz,cdcl3)δ8.70(d,j=2.4hz,1h),8.56-8.43(m,1h),8.19(d,j=8.1hz,1h),8.11(d,j=7.4hz,1h),7.95(d,j=5.2hz,1h),7.89(d,j=10.0hz,1h),7.73-7.59(m,2h),7.34(dd,j=8.0,3.9hz,1h),7.22(t,j=7.7hz,1h),7.14(dd,j=17.5,8.5hz,2h),6.40(d,j=7.9hz,1h);
核磁碳谱图如附图9所示,具体数据为13cnmr(126mhz,cdcl3)δ161.26,151.41,144.43,139.90,136.35,135.78,132.34,132.26,130.51,129.76,129.49,126.75,123.27,122.56,121.96,117.00,116.23,116.05,107.93,107.74;
hrms(esi)calcdforc22h13fn2o:[m+h]+340.3498,found:340.1012.
上述数据说明本实施例成功合成了9-氟-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料4-氟-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.5ml4-氟苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到4.1克4-氟-n-(喹啉-8-基)苯甲酰胺,产率为78%。
原料4-氟-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=117~118℃;
1hnmr(500mhz,cdcl3):δ10.86(s,1h),8.87(d,j=8.0hz,1h),8.86-8.81(m,1h),8.20-8.14(m,1h),8.12-8.05(m,2h),7.61-7.55(m,1h),7.55-7.50(m,1h),7.49-7.43(m,1h),7.24-7.18(m,2h);
13cnmr(125mhz,cdcl3):δ165.32(d,j=252.8hz),164.64,148.61,139.04,136.75,134.73,131.68,130.09(d,j=8.8hz),128.30,127.74,122.18,122.04,116.87,116.15(d,j=21.9hz);
hrms(esi)calcdforc16h12fn2o:[m+h]+267.0928,found:267.0927。
上述数据说明成功合成了原料4-氟-n-(喹啉-8-基)苯甲酰胺。
实施例5:9-氯-5-(喹啉-8-基)菲啶-6(5h)-酮
将282mg(1mmol)4-氯-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得232mg黄色固体,产率为65%。
所得产物的各项表征数据如下:
m.p.=244~245℃;
核磁氢谱图如附图10所示,具体数据为1hnmr(500mhz,cdcl3)δ8.79(dd,j=4.2,1.7hz,1h),8.51(d,j=8.5hz,1h),8.34(d,j=1.9hz,1h),8.27(ddd,j=16.0,8.1,1.5hz,2h),8.07-8.02(m,1h),7.79-7.72(m,2h),7.56(dd,j=8.5,1.9hz,1h),7.44(dd,j=8.3,4.2hz,1h),7.28-7.18(m,3h),6.49(dd,j=8.2,1.1hz,1h);
核磁碳谱图如附图11所示,具体数据为13cnmr(126mhz,cdcl3)δ161.33,151.44,144.39,139.89,139.49,136.36,135.91,135.74,130.90,130.48,129.78,129.76,129.54,128.32,126.76,124.34,123.17,122.67,121.99,121.83,118.01,117.02;
hrms(esi)calcdforc22h13cln2o:[m+h]+356.8044,found:356.0816.
上述数据说明本实施例成功合成了9-氯-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料4-氯-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.7ml4-氯苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到4.8克4-氯-n-(喹啉-8-基)苯甲酰胺,产率为85%。
4-氯-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=115~116℃;
1hnmr(500mhz,cdcl3):δ10.59(brs,1h),8.84(dd,j=7.4,1.5hz,1h),8.74(dd,j=4.2,1.6hz,1h),8.05(dd,j=8.3,1.6hz,1h),7.95-7.90(m,2h),7.52-7.34(m,5h);
13cnmr(125mhz,cdcl3):δ163.98,148.21,138.53,137.97,136.25,134.20,133.33,128.90,128.58,127.83,127.27,121.79,121.64,116.45;
hrms(esi)calcdforc16h12cln2o:[m+h]+283.0633,found:283.0635.
上述数据说明成功合成了原料4-氯-n-(喹啉-8-基)苯甲酰胺。
实施例6:9-(三氟甲基)-5-(喹啉-8-基)菲啶-6(5h)-酮
将316mg(1mmol)4-三氟甲基-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得316mg黄色固体,产率为81%。
所得产物的各项表征数据如下:
m.p.=265~266℃;
1hnmr(500mhz,cdcl3)δ8.79(dd,j=4.2,1.7hz,1h),8.70(d,j=8.3hz,1h),8.63(d,j=15.0hz,1h),8.37(dd,j=8.0,1.4hz,1h),8.30(dd,j=8.3,1.7hz,1h),8.10-7.96(m,1h),7.83(dd,j=8.3,1.1hz,1h),7.80-7.73(m,2h),7.45(dd,j=8.3,4.2hz,1h),7.30(tt,j=6.9,3.4hz,1h),7.28-7.21(m,1h),6.53(dd,j=8.3,1.0hz,1h);
13cnmr(126mhz,cdcl3)δ161.04,151.49,144.30,139.81,136.39,135.60,134.80,134.47,134.21,130.42,130.20,130.01,129.82,129.66,128.31,126.78,124.97,124.03,124.00,123.22,122.89,122.79,122.07,119.38,119.35,118.21,117.13;
hrms(esi)calcdforc23h13f3n2o:[m+h]+390.3573,found:390.3570.
上述数据说明本实施例成功合成了9-(三氟甲基)-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料4-三氟甲基-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加3.9ml4-三氟甲基-苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到5.4克4-三氟甲基-n-(喹啉-8-基)苯甲酰胺,产率为85%。
4-三氟甲基-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=156~157℃;
1hnmr(500mhz,cdcl3):δ10.80(s,1h),8.92(dd,j=7.2,1.7hz,1h),8.86(dd,j=4.2,1.7hz,1h),8.25-8.16(m,3h),7.82(d,j=8.1hz,2h),7.65-7.56(m,2h),7.51(dd,j=8.3,4.2hz,1h);
13cnmr(125mhz,cdcl3):δ164.14,148.42,138.85,138.58,136.52,134.26,133.54(q,jc-f=32.9hz),128.03,127.81,127.50,125.92(q,jc-f=3.7hz),123.73(q,jc-f=272.2hz),122.25,121.86,116.80;
hrms(esi)calcdforc17h12f3n2o:[m+h]+317.0896,found317.0880.
上述数据说明成功合成了原料4-三氟甲基-n-(喹啉-8-基)苯甲酰胺。
实施例7:9-溴-5-(喹啉-8-基)菲啶-6(5h)-酮
将327mg(1mmol)4-溴-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得249mg黄色固体,产率为62%。
所得产物的各项表征数据如下:
m.p.=264~266℃;
1hnmr(500mhz,cdcl3)δ8.79(dd,j=4.2,1.7hz,1h),8.51(d,j=1.8hz,1h),8.42(d,j=8.5hz,1h),8.27(ddd,j=16.0,8.1,1.6hz,2h),8.05(dd,j=6.4,3.3hz,1h),7.80-7.73(m,2h),7.71(dd,j=8.5,1.8hz,1h),7.44(dd,j=8.3,4.2hz,1h),7.30-7.17(m,3h),6.49(dd,j=8.3,1.2hz,1h);
13cnmr(126mhz,cdcl3)δ161.44,151.43,144.35,139.84,136.37,136.04,135.71,131.13,130.90,130.47,129.76,129.54,128.20,126.76,124.94,124.69,123.15,122.69,121.99,117.88,117.00;
hrms(esi)calcdforc22h13brn2o:[m+h]+401.2554,found:401.2552.
上述数据说明本实施例成功合成了9-溴-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料4-溴-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.9ml4-溴苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到4.3克4-溴-n-(喹啉-8-基)苯甲酰胺,产率为65%。
原料4-溴-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=113~135℃;
1hnmr(400mhz,cdcl3)δ10.71(s,1h),8.90(dd,j=7.3,1.4hz,1h),8.85(dd,j=4.2,1.5hz,1h),8.19(dd,j=8.3,1.5hz,1h),7.96(t,j=6.5hz,2h),7.73–7.64(m,2h),7.63–7.52(m,2h),7.48(dd,j=8.3,4.2hz,1h);
13cnmr(101mhz,cdcl3)δ164.39,148.32,138.69,136.43,134.30,133.97,132.35,132.01,131.88,128.87,127.97,127.43,126.58,121.89,121.74,116.60;
hrms(esi)calcdforc16h11brn2o:[m+h]+327.1753,found:327.1743.
上述数据说明成功合成了原料4-溴-n-(喹啉-8-基)苯甲酰胺。
实施例8:9-乙基-5-(喹啉-8-基)菲啶-6(5h)-酮
将276mg(1mmol)4-乙基-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得182mg黄色固体,产率为52%。
所得产物的各项表征数据如下:
m.p.=207~209℃;
1hnmr(500mhz,cdcl3)δ8.78(dd,j=4.2,1.7hz,1h),8.50(d,j=8.1hz,1h),8.38-8.33(m,1h),8.27(dd,j=8.3,1.7hz,1h),8.19(s,1h),8.05–8.00(m,1h),7.76(t,j=5.0hz,2h),7.48(s,1h),7.42(dd,j=8.3,4.2hz,1h),7.25(d,j=7.2hz,2h),7.18(s,1h),6.48(dd,j=8.3,0.9hz,1h),2.91(q,j=7.6hz,2h),1.39(t,j=7.6hz,3h);
13cnmr(126mhz,cdcl3)δ151.34,149.44,144.60,139.64,136.28,136.19,134.46,130.62,129.76,129.30,129.21,128.84,128.24,126.74,123.93,122.98,122.29,121.87,120.75,119.18,116.84,29.52,15.52,0.99;
hrms(esi)calcdforc24h18n2o:[m+h]+350.4125,found:350.4413.
上述数据说明本实施例成功合成了9-乙基-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料4-乙基-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.8ml4-乙基苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到3.6克4-乙基-n-(喹啉-8-基)苯甲酰胺,产率为65%。
原料4-乙基-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=68~70℃;
1hnmr(400mhz,cdcl3)δ10.65(s,1h),8.86(d,j=7.2hz,1h),8.76(s,1h),8.09(d,j=8.0hz,1h),7.93(d,j=7.4hz,2h),7.51(s,1h),7.45(s,1h),7.41-7.35(m,1h),7.29(d,j=7.4hz,2h),2.66(d,j=7.4hz,2h),1.21(t,j=7.3hz,3h);
13cnmr(101mhz,cdcl3)δ165.54,148.60,148.28,136.42,134.71,132.58,128.33,128.01,127.51,127.44,121.70,121.57,116.46,28.90,15.42;
hrms(esi)calcdforc18h16n2o:[m+h]+276.3324,found:276.3320.
上述数据说明成功合成了原料4-乙基-n-(喹啉-8-基)苯甲酰胺。
实施例9:8-乙基-5-(喹啉-8-基)菲啶-6(5h)-酮
将262mg(1mmol)3-甲基-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得188mg黄色固体,产率为56%
所得产物的各项表征数据如下:
m.p.=260~262℃;
1hnmr(500mhz,cdcl3)δ8.79(dd,j=4.2,1.7hz,1h),8.38(d,j=0.5hz,1h),8.33-8.24(m,3h),8.09-7.99(m,1h),7.85-7.71(m,2h),7.68-7.59(m,1h),7.47-7.37(m,1h),7.30-7.20(m,1h),7.16(ddd,j=8.5,7.2,1.4hz,1h),6.48(dd,j=8.4,1.0hz,1h),2.60-2.43(m,3h);
13cnmr(126mhz,cdcl3)δ162.19,151.50,144.72,139.31,138.20,136.44,136.37,134.21,132.08,130.69,129.92,129.48,128.97,128.66,126.90,125.97,122.95,122.53,122.02,119.39,116.93,21.53;
hrms(esi)calcdforc23h16n2o:[m+h]+336.1263found:336.1233.
上述数据说明本实施例成功合成了8-乙基-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料3-甲基-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.8ml3-甲基苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到4.1克3-甲基-n-(喹啉-8-基)苯甲酰胺,产率为78%。
原料3-甲基-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=49~50℃;
1hnmr(500mhz,cdcl3):δ10.63(s,1h),8.93-8.90(m,1h),8.75-8.74(m,1h),8.04-8.01(m,1h),7.85-7.82(m,2h),7.52-7.48(t,j=8.0hz,,1h),7.43-7.40(m,1h),7.38-7.36(m,1h),7.34-7.29(m,3h),2.41(s,3h);
13cnmr(125mhz,cdcl3):δ165.32,148.04,138.51,138.48,136.09,134.94,134.47,132.42,128.48,127.81,127.75,127.16,124.02,121.45,116.21,21.27;
hrms(esi)calcdforc17h14n2o:[m+h]+262.1106,found262.2311.
上述数据说明成功合成了原料3-甲基-n-(喹啉-8-基)苯甲酰胺。
实施例10:8-氯-5-(喹啉-8-基)菲啶-6(5h)-酮
将282mg(1mmol)3-氯-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得218mg黄色固体,产率为61%。
所得产物的各项表征数据如下:
m.p.=295~298℃;
1hnmr(500mhz,dmso)δ8.72(t,j=6.6hz,2h),8.57(d,j=7.8hz,2h),8.26(d,j=7.7hz,2h),8.01(dd,j=8.7,2.1hz,1h),7.94(d,j=6.8hz,1h),7.87(t,j=7.7hz,1h),7.61(dd,j=8.3,4.1hz,1h),7.31(t,j=5.5hz,2h),6.44–6.33(m,1h);
13cnmr(126mhz,dmso)δ159.64,151.30,143.59,139.18,136.73,135.12,133.26,133.10,132.82,130.78,130.01,129.72,129.39,127.07,126.98,126.77,125.35,123.79,122.79,122.31,117.51,116.59;
hrms(esi)calcdforc22h13cln2o:[m+h]+356.0716,found:356.0722.
上述数据说明本实施例成功合成了8-氯-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料3-氯-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加2.7ml3-氯苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到4.5克3-氯-n-(喹啉-8-基)苯甲酰胺,产率为80%。
原料3-氯-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=87~88℃;
1hnmr(400mhz,cdcl3)δ10.69(d,j=14.0hz,1h),8.95-8.77(m,2h),8.24-8.11(m,1h),8.05(dd,j=6.9,1.6hz,1h),7.94(t,j=9.4hz,1h),7.65-7.40(m,5h);
13cnmr(101mhz,cdcl3)δ148.40,136.88,136.43,135.00,131.86,130.07,127.94,127.70,127.42,127.37,125.26,125.22,122.01,121.77,116.71,116.67;
hrms(esi)calcdforc16h11cln2o:[m+h]+282.0560,found:282.0552.
上述数据说明成功合成了原料3-氯-n-(喹啉-8-基)苯甲酰胺。
实施例11:8-(三氟甲基)-5-(喹啉-8-基)菲啶-6(5h)-酮
将316mg(1mmol)3-三氟甲基-n-(喹啉-8-基)苯甲酰胺和301mg(1.5mmol)邻溴苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂)得293mg黄色固体,产率为75%。
所得产物的各项表征数据如下:
m.p.=212~213℃;
1hnmr(500mhz,cdcl3)δ8.87(s,1h),8.78(dd,j=4.2,1.7hz,1h),8.47(d,j=8.6hz,1h),8.36-8.31(m,1h),8.29(dd,j=8.3,1.7hz,1h),8.06(dd,j=7.0,2.7hz,1h),8.00(dd,j=8.6,1.9hz,1h),7.81-7.72(m,2h),7.43(dd,j=8.3,4.2hz,1h),7.31-7.22(m,2h),6.57-6.47(m,1h);
13cnmr(126mhz,cdcl3)δ161.24,151.55,144.42,140.21,137.35,136.55,135.66,130.56,130.45,129.96,129.82,128.98,128.95,126.90,126.85,126.82,126.16,123.75,123.01,122.18,118.14,117.24;
hrms(esi)calcdforc23h13f3n2o:[m+h]+390.3573,found:390.3666.
上述数据说明本实施例成功合成了8-(三氟甲基)-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料3-(三氟甲基)-n-(喹啉-8-基)苯甲酰胺的合成采用下述步骤:在250ml圆底烧瓶中加入2.88g8-氨基喹啉(20mmol),5.5ml三乙胺(40mmol)和干燥的二氯甲烷中(40ml)并搅拌10min,待溶解完全后置于冰浴中,用恒压滴液漏斗慢慢滴加3.1ml3-(三氟甲基)-苯甲酰氯(21mmol)的二氯甲烷溶液(10ml),上述反应液在冰浴下继续搅拌半小时后慢慢升温至室温。跟踪反应情况,待板上只有一个点时,慢慢加入冰水淬灭反应。将有机相分离,并用二氯甲院萃取水相两次,收集有机相,用无水mgso4干燥,用旋转蒸发仪蒸除溶剂,柱层析分离(乙酸乙酯:石油醚=1:8做洗脱剂),得到5.2克3-三氟甲基-n-(喹啉-8-基)苯甲酰胺,产率为82%。
3-三氟甲基-n-(喹啉-8-基)苯甲酰胺的各项表征数据如下:
m.p.=88~89℃;
1hnmr(400mhz,cdcl3)δ10.77(s,1h),8.91(d,j=7.1hz,1h),8.86(d,j=1.7hz,1h),8.35(s,1h),8.25(d,j=7.7hz,1h),8.20(d,j=8.2hz,1h),7.84(d,j=7.7hz,1h),7.69(t,j=7.7hz,1h),7.59(q,j=8.4hz,2h),7.49(dd,j=7.9,3.9hz,1h);
13cnmr(101mhz,cdcl3)δ163.87,148.44,138.67,136.44,135.95,134.11,130.22,129.38,128.39,128.35,127.96,127.38,124.57,124.53,122.13,121.80,116.74;
hrms(esi)calcdforc17h11f3n2o:[m+h]+316.0823,found316.1232.
上述数据说明成功合成了原料3-三氟甲基-n-(喹啉-8-基)苯甲酰胺。
实施例12:2-氯-8-甲基-5-(喹啉-8-基)菲啶-6(5h)-酮
将262mg(1mmol)3-甲基-n-(喹啉-8-基)苯甲酰胺和352mg(1.5mmol)2-溴-5-氯苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:3做洗脱剂)得274mg黄色固体,产率为74%。
所得产物的各项表征数据如下:
m.p.=247~248℃;
1hnmr(500mhz,cdcl3):δ8.77(dd,j=1.6,4.0hz,1h),8.35(d,j=0.4hz,1h),8.26(dd,j=1.6,8.4hz,1h),8.23(d,j=2.0hz,1h),8.18(d,j=8.4hz,1h),8.03(dd,j=4.4,5.2hz,1h),7.72-7.77(m,2h),7.64(d,j=8.4hz,1h),7.42(dd,j=4.0,8.4hz,1h),7.08(dd,j=2.4,8.8hz,1h),6.39(d,j=8.8hz,1h),2.52(s,3h);
13cnmr(100mhz,cdcl3):δ161.9,151.6,144.6,139.1,137.9,136.5,136.0,134.4,130.9,130.7,130.0,129.7,129.1,128.5,128.2,126.9,126.2,122.7,121.13,122.12,120.7,118.3,21.6;
hrms(esi)calcdforc23h16cln2o:[m+h]+371.0946,found:371.0951.
上述数据说明本实施例成功合成了2-氯-8-甲基-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料3-甲基-n-(喹啉-8-基)苯甲酰胺的合成同实例9一致。
实施例13:2-氟-8-甲基-5-(喹啉-8-基)菲啶-6(5h)-酮
将262mg(1mmol)3-甲基-n-(喹啉-8-基)苯甲酰胺和327mg(1.5mmol)2-溴-5-氟苯硼酸,247mg(2mmol)催化剂cu(oac)2,651mg(2mmol)碱cs2co3,3ml溶剂二甲基亚砜(dmso)在氮气环境下加入30ml封管中。然后将封管放入100℃油浴中反应4h。反应结束后,将反应液冷却至室温,加入80ml乙酸乙酯,快速柱层析(乙酸乙酯做洗脱剂),合并有机层,有机层经水洗,无水硫酸钠干燥,过滤,滤液减压蒸馏后经硅胶柱层析分离(乙酸乙酯:石油醚=1:3做洗脱剂)得184mg黄色固体,产率为52%。
所得产物的各项表征数据如下:
m.p.=234~235℃;
1hnmr(400mhz,cdcl3):δ8.78(dd,j=1.6,4.0hz,1h),8.36(s,1h),8.27(dd,j=1.6,8.4hz,1h),8.15(d,j=8.4hz,1h),8.03(dd,j=3.6,6.0hz,1h),7.94(dd,j=2.8,10.0hz,1h),7.72-7.78(m,2h),7.64(dd,j=1.6,8.4hz,1h),7.42(dd,j=4.0,8.4hz,1h),6.87(ddd,j=2.0,8.0,9.2hz,1h),6.42(dd,j=4.8,9.2hz,1h),2.53(s,3h);
13cnmr(100mhz,cdcl3):δ161.8,158.7(d,j=241.3hz),151.6,144.7,139.0,136.5,136.3,135.8(d,j=1.4hz),134.3,131.2(d,j=2.8hz),130.7,130.0,129.6,129.2,126.9,126.2,122.2,122.1,120.7(d,j=8.1hz),118.4(d,j=8.1hz),116.0(d,j=23.7hz),108.9(d,j=23.7hz),21.6;
19fnmr(376mhz,cdcl3)δ-120.93;
hrms(esi)calcdforc23h16fn2o:[m+h]+355.1241,found355.1244
上述数据说明本实施例成功合成了2-氟-8-甲基-5-(喹啉-8-基)菲啶-6(5h)-酮。
原料3-甲基-n-(喹啉-8-基)苯甲酰胺的合成同实例9一致。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!