一种化学交联剂及其制备和应用的制作方法
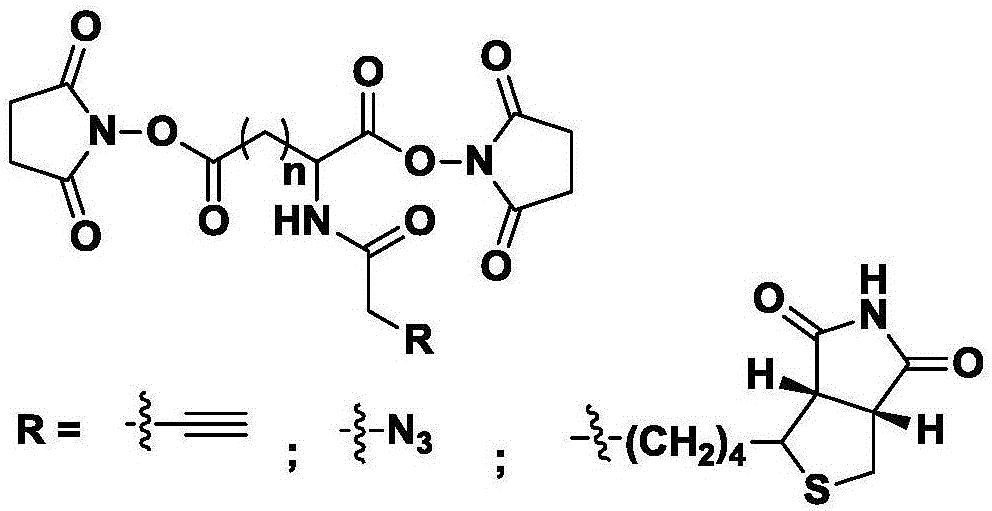
本发明涉及一种氨基酸骨架结构、同时具有同位素标记及富集功能的化学交联剂及其制备方法和应用。本发明交联剂采用不同碳链长度的氨基酸为骨架结构,具体包括两种天然氨基酸:天冬氨酸、谷氨酸;三种非天然氨基酸:2-氨基-1,6-己二酸、2-氨基-1,7-庚二酸、2-氨基-1,8-辛二酸。氨基酸骨架引入同位素标记,包括氘、碳13或氮15的一种或两种以上。交联剂修饰化学富集基团,包括炔基、叠氮或生物素的一种。该发明属于有机合成技术领域。
背景技术:
近年来,化学交联剂结合质谱技术兴起,成为一种研究蛋白质复合物结构以及蛋白质-蛋白质相互作用的有力工具。该方法相比酵母双杂交、免疫共沉淀、蛋白质结晶x射线衍射等传统方法(trendsbiotechnol,2016,34,825-834.;journalofcellularbiochemistry,2016,117,2109-2117.)具有分析迅速、灵敏度高、通量高、可处理细胞裂解液、完整细胞及组织蛋白样品等优点,因而成为持续增长的新的研究热点(massspectrometryreviews,2010,29,862-876;analyticalchemistry,2016,88,7930-7937;journalofproteomeresearch,2017,1,722.)。
化学交联剂的设计与合成,为化学交联剂结合质谱技术提供重要的技术支撑。一般而言,化学交联剂包含两个反应活性基团以及合理的间隔臂长。目前反应活性基团最常采用的是琥珀酰亚胺酯结构,可以特异性地与蛋白质或多肽赖氨酸末端或n端氨基反应,反应条件温和、高效。交联反应后的蛋白质再经过酶解即可得到交联肽。由于交联肽在常规肽(未交联的多肽)中占有率小,丰度低,为实现交联肽的高通量分析,人们在交联剂的设计过程中引入富集基团。目前较为常见的富集基团包括炔基、叠氮以及生物素基团(journalofproteomeresearch,2009,8,3702-3711;journaloftheamericanchemicalsociety,2003,9,241625;molecular&cellularproteomics,2014,13,3533.)。其中炔基可以与含有叠氮的磁球发生点击化学反应,实现交联肽高效富集;同理,叠氮可以与含有炔基的磁球发生点击化学反应,实现交联肽高效富集;生物素单元可以采用含有链霉亲和素的磁球,实现交联肽高效富集。
细胞或组织蛋白原位交联可以提供更真实的蛋白结构及蛋白-蛋白相互作用信息,是化学交联质谱技术的热点研究领域(currentopinioninchemicalbiology,2019,48,8;methods,2018,15,144;cellchemicalbiology,2017,25,1)。为了实现化学交联剂的穿膜特性,将化学交联剂设计成电中性、疏水结构是主要的方法,如报道较多的disuccinimidylsuberate(简称dss)、disuccinimidylsulfoxide(简称dsso)均具有很好的穿膜特性。美国华盛顿大学的bruce课题组在交联剂的设计方面做出了突出的工作,他所设计的化学交联剂(统一命名为pir)均以多肽链为骨架结构,得到了较好的交联效果(molecularbiosystems,2010,6,939;naturecommunications,2016,7,13414;pnas,2017,114,1732.)。然而,由于交联剂具有大的骨架结构,引入更多的空间位阻,不利于蛋白内部的化学交联。2018年,heck课题组利用小尺寸的dsso交联剂应用于细胞线粒体蛋白交联,得到的交联肽数目、交联蛋白数目均多于pir交联剂,说明尺寸小的交联剂具有更小的位阻、更灵活的优势,因而也是未来交联剂的发展趋势(molecular&cellularprotomics,2018,17,216.)。
化学交联剂引入同位素标记,可以实现化学交联质谱技术与定量蛋白组学技术相结合,获取细胞内蛋白的时空动态变化信息,为生命科学领域提供更有力的技术支撑(currentopinioninchemicalbiology,2019,48,8)。
技术实现要素:
基于上述化学交联剂的研究现状及设计原则,本发明设计并合成了一种氨基酸骨架结构、具有同位素标记及富集功能的化学交联剂。本发明氨基酸骨架具有不同的碳链长度,具体包括两种天然氨基酸:天冬氨酸、谷氨酸;三种非天然氨基酸:2-氨基-1,6-己二酸、2-氨基-1,7-庚二酸、2-氨基-1,8-辛二酸。氨基酸骨架引入同位素标记,包括氘、碳13或氮15的一种或两种以上,实现化学交联质谱技术与定量蛋白组学技术相结合。氨基酸末端羧基引入琥珀酰亚胺酯活性基团,能够高效温和地和蛋白、多肽赖氨酸末端或n端氨基发生交联反应,实现蛋白质的快速交联。另外,针对交联肽在常规肽中占有率小、丰度低的问题,本发明在交联剂中引入富集基团,包括炔基、叠氮或生物素,实现交联肽的富集,提高其在质谱中的响应,实现交联肽的高通量、高灵敏度分析。化学交联剂具有电中性、疏水结构,满足交联剂的穿膜要求,能够实现细胞内蛋白原位化学交联,获取高可信度的蛋白三维结构以及蛋白-蛋白相互作用信息。
本发明提供的一种氨基酸骨架、具有同位素标记及富集功能的化学交联剂,结构如下:
氨基酸骨架包括由两个酯羰基及之间的碳链以及氨基部分构成。
其中,n取值范围为1-5,对应不同碳链长度的氨基酸骨架;氨基酸骨架引入不同的同位素标记,包括氘、碳13或氮15的一种或两种以上;r为化学富集基团,包括炔基、叠氮或生物素的一种。
具体包含15种结构,如下:
天冬氨酸骨架、不同富集基团的化学交联剂,具有如下结构:
谷氨酸骨架、不同富集基团的化学交联剂,具有如下结构:
2-氨基-1,6-己二酸骨架、不同富集基团的化学交联剂,具有如下结构:
2-氨基-1,7-庚二酸骨架、不同富集基团的化学交联剂,具有如下结构:
2-氨基-1,8-辛二酸骨架、不同富集基团的化学交联剂,具有如下结构:
其中,上述15种化学交联剂均可以在氨基酸骨架上引入同位素标记,包括氘、碳13、氮15的一种或两种以上。
本发明提供了化学交联剂的制备方法,具体步骤如下:
第一步,富集基团酰氯试剂的制备。将具有富集基团的羧酸溶于无水二氯甲烷中,加入0.01-0.1mldmf,反应液降温至0-5℃,取乙二酰氯滴加至反应液,乙二酰氯与富集基团的羧酸摩尔比控制在(1-1.5):1,滴毕,反应液升温至15-25℃,继续反应1-2h,即制得具有富集基团的酰氯试剂;
第二步,修饰富集基团的二羧酸化合物的制备。将不同碳链长度或同位素标记的氨基酸溶于饱和的碳酸氢钠中,氨基酸与碳酸氢钠摩尔比控制在1:(3-5),反应液降温至0-5℃,滴加步骤一新制备的具有富集基团的酰氯试剂,氨基酸与富集基团酰氯试剂摩尔比控制在1:(1.1-1.3),滴毕,反应液升温至15-25℃,继续反应1-2h,反应毕,饱和氢氧化钠溶液调反应液ph为10-12,乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为1-3,乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,即制得修饰富集基团的二羧酸化合物;
第三步,目标化学交联剂的合成。将修饰富集基团的二羧酸化合物、edci与nhs置于无水二氯甲烷中,三者的摩尔比控制在1:(2.2-3.0):(2.2-3.0),20-25℃搅拌12-24h,反应毕,减压蒸干有机溶剂,将粗产物溶于dmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,合并产物流出液,35-40℃真空冻干,即制得不同碳链长度氨基酸骨架具有同位素标记富集型的化学交联剂。
本发明提供的交联剂是一种以不同碳链长度的氨基酸为骨架结构、具有同位素标记及富集功能的化学交联剂。本发明化学交联剂应用领域是结构蛋白质组学,为细胞内原位蛋白复合体的规模化分析、蛋白质的空间结构解析、蛋白质-蛋白质相互作用解析以及蛋白质时空动态变化分析提供重要的技术支撑。
与现有化学交联剂相比,本发明交联剂具有如下优点:
1.以氨基酸为骨架结构,尺寸小,交联位阻小,有利于提高交联反应效率,获取更多的交联信息。
2.具有富集基团,包括炔基、叠氮或生物素基团,可以实现交联肽的有效富集,解决样品复杂、交联肽丰度低的问题,提高交联肽在质谱中的响应,实现高通量、高灵敏度分析。
3.具有同位素标记,包括氘、碳13、氮15的一种或几种,实现化学交联质谱技术与定量蛋白组学技术相结合,获取细胞内蛋白质时空动态变化的信息。
4.具有电中性、疏水结构,交联剂具有透膜特性,能够穿过细胞膜到达细胞质及各个亚细胞器,实现细胞内蛋白原位化学交联,获取高可信度的蛋白三维结构以及蛋白-蛋白相互作用信息。
附图说明
图1:化学交联剂结构通式;
图2:化学交联剂的合成路线;
图3:天冬氨酸骨架交联剂的具体结构式;
图4:谷氨酸骨架交联剂的具体结构式;
图5:2-氨基-1,6-己二酸骨架交联剂的具体结构式;
图6:2-氨基-1,7-庚二酸骨架交联剂的具体结构式;
图7:2-氨基-1,8-辛二酸骨架交联剂的具体结构式。
具体实施方式
以下结合具体实施例,对本发明作进一步说明。
实施例1
本实施例公开了一种天冬氨酸骨架、具有炔基富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,3-丁炔酰氯的制备(参考文献chemicalcommunications,2012,48,6496.)。将3-丁炔酸(1.0g,12mmol)溶于15ml无水二氯甲烷中,加入0.01-0.1mldmf,反应液降温至0℃,取乙二酰氯(1.64g,13mmol,1.1ml)滴加至反应液。滴毕,反应液升温至25℃,继续反应1h,即制得3-丁炔酰氯的二氯甲烷溶液;
第二步,2-丁炔酰胺基-1,4-丁二酸的制备。将l-天冬氨酸(1.34g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加3-丁炔酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.45g,收率73%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.65-2.60(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,76.3,71.2,53.4,36.1,24.2;hr-ms(c8h9no5):理论值:199.0481,测定值[m+h]:200.2018。
第三步,2-丁炔酰胺基-1,4-丁二琥珀酰亚胺酯的制备。将2-丁炔酰胺基-1,4-丁二酸(1g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间22-24min,合并产物流出液,40℃真空冻干24h,得到白色固体1.63g,收率83%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.80(s,8h),2.65-2.60(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,76.3,71.2,53.4,36.1,24.2;hr-ms(c16h15n3o9):理论值:393.0808,测定值[m+h]:394.3123。
实施例2
本实施例公开了一种天冬氨酸骨架、具有叠氮富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,2-叠氮乙酰氯的制备。将2-叠氮乙酸(1.21g,12mmol)溶于15ml无水二氯甲烷中,加入0.01-0.1mldmf,反应液降温至0℃,取乙二酰氯(1.64g,13mmol,1.1ml)滴加至反应液。滴毕,反应液升温至25℃,继续反应1h,即制得2-叠氮乙酰氯的二氯甲烷溶液。
第二步,2-叠氮乙酰胺基-1,4-丁二酸的制备。将l-天冬氨酸(1.34g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加2-叠氮乙酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.47g,收率68%。1hnmr(400mhz,d2o,ppm)δ5.83(s,2h),4.76(s,1h),2.65-2.60(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,53.4,36.1,24.2;hr-ms(c6h8n4o5):理论值:216.0495,测定值[m+h]:217.2214。
第三步,2-叠氮乙酰胺基-1,4-丁二琥珀酰亚胺酯的制备。将2-叠氮乙酰胺基-1,4-丁二酸(1.08g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间18-20min,合并产物流出液,40℃真空冻干24h,得到白色固体1.63g,收率83%。1hnmr(400mhz,dmso-d6,ppm)δ5.83(s,2h),4.76(s,1h),2.80(s,8h),2.65-2.60(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,36.1,24.2;hr-ms(c14h14n6o9):理论值:410.0822,测定值[m+h]:411.3521。
实施例3
本实施例公开了一种天冬氨酸骨架、具有生物素结构的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,5-生物素酰氯的制备。将生物素(2.93g,12mmol)溶于30ml无水二氯甲烷中,加入0.01-0.1mldmf,反应液降温至0℃,取乙二酰氯(1.64g,13mmol,1.1ml)滴加至反应液。滴毕,反应液升温至25℃,继续反应1h,即制得5-生物素酰氯的二氯甲烷溶液;
第二步,5-生物素酰胺基-1,4-丁二酸的制备。将l-天冬氨酸(1.34g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加5-生物素酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体3.02g,收率84.2%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.65-2.60(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,d2o,ppm)δ179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,27.4,25.5,23.1;hr-ms(c14h21n3o6s):理论值:359.1151,测定值[m+h]:360.3256。
第三步,5-生物素酰胺基-1,4-丁二琥珀酰亚胺酯的制备。将5-生物素酰胺基-1,4-丁二酸(1.8g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度40-70,30min,产品出峰时间25-27min,合并产物流出液,40℃真空冻干24h,得到白色固体1.85g,收率67.8%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.8(s,8h),2.65-2.60(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ184.3,179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,32.4,27.4,25.5,23.1;hr-ms(c22h27n5o10s):理论值:553.1479,测定值[m+h]:554.4371。
实施例4
本实施例公开了一种谷氨酸骨架、具有炔基富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,3-丁炔酰氯的制备。同实施例1第一步。
第二步,2-丁炔酰胺基-1,5-戊二酸的制备。将l-谷氨酸(1.47g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加3-丁炔酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.62g,收率76%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.72(d,j=4.8hz,2h),2.65-2.60(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,76.3,71.2,53.4,36.1,28.1,24.2;hr-ms(c9h11no5):理论值:213.0637,测定值[m+h]:214.2418。
第三步,2-丁炔酰胺基-1,5-戊二琥珀酰亚胺酯的制备。将2-丁炔酰胺基-1,5-戊二酸(1.07g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间22-24min,合并产物流出液,40℃真空冻干24h,得到白色固体1.42g,收率78%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.80(s,8h),2.72(d,j=4.8hz,2h),2.65-2.60(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,76.3,71.2,53.4,36.1,28.1,24.2;hr-ms(c17h17n3o9):理论值:407.0965,测定值[m+h]:408.2913。
实施例5
本实施例公开了一种谷氨酸骨架、具有叠氮富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,2-叠氮乙酰氯的制备。同实施例2第一步。
第二步,2-叠氮乙酰胺基-1,5-戊二酸的制备。将l-谷氨酸(1.47g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加2-叠氮乙酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.36g,收率59.2%。1hnmr(400mhz,d2o,ppm)δ5.83(s,2h),4.76(s,1h),2.72(d,j=4.8hz,2h),2.65-2.60(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,53.4,36.1,28.1,24.2;hr-ms(c7h10n4o5):理论值:230.0651,测定值[m+h]:231.2561。
第三步,2-叠氮乙酰胺基-1,5-戊二琥珀酰亚胺酯的制备。将2-叠氮乙酰胺基-1,5-戊二酸(1.15g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间18-20min,合并产物流出液,40℃真空冻干24h,得到白色固体1.59g,收率75%。1hnmr(400mhz,dmso-d6,ppm)δ5.83(s,2h),4.76(s,1h),2.80(s,8h),2.72(d,j=4.8hz,2h),2.65-2.60(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,36.1,28.1,24.2;hr-ms(c15h16n6o9):理论值:424.0979,测定值[m+h]:425.3521。
实施例6
本实施例公开了一种谷氨酸骨架、具有生物素结构的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,5-生物素酰氯的制备。同实施例3第一步。
第二步,5-生物素酰胺基-1,5-戊二酸的制备。将l-谷氨酸(1.47g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加5-生物素酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体2.05g,收率55.1%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.72(d,j=6.4hz,2h),2.65-2.60(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,d2o,ppm)δ179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,28.1,27.4,25.5,23.1;hr-ms(c15h23n3o6s):理论值:373.1308,测定值[m+h]:374.3419。
第三步,5-生物素酰胺基-1,5-戊二琥珀酰亚胺酯的制备。将5-生物素酰胺基-1,5-戊二酸(1.87g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度40-70,30min,产品出峰时间25-27min,合并产物流出液,40℃真空冻干24h,得到白色固体2.01g,收率70.9%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.81(s,8h),2.72(d,j=6.4hz,2h),2.65-2.60(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ184.3,179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,32.4,28.1,27.4,25.5,23.1;hr-ms(c23h29n5o10s):理论值:567.1635,测定值[m+h]:568.4317。
实施例7
本实施例公开了一种2-氨基-1,6-己二酸骨架、具有炔基富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,3-丁炔酰氯的制备。同实施例1第一步。
第二步,2-丁炔酰胺基-1,6-己二酸的制备。将2-氨基-1,6-己二酸(1.61g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加3-丁炔酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.76g,收率76%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.78(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.65-2.60(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,76.3,71.2,53.4,36.1,30.1,28.1,24.2;hr-ms(c10h13no5):理论值:227.0794,测定值[m+h]:228.4104。
第三步,2-丁炔酰胺基-1,6-己二琥珀酰亚胺酯的制备。将2-丁炔酰胺基-1,6-己二酸(1.14g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间22-24min,合并产物流出液,40℃真空冻干24h,得到白色固体1.52g,收率72.2%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.80(s,8h),2.79(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.65-2.60(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,76.3,71.2,53.4,36.1,30.3,28.1,24.2;hr-ms(c18h19n3o9):理论值:421.1121,测定值[m+h]:422.4512。
实施例8
本实施例公开了一种2-氨基-1,6-己二酸骨架、具有叠氮富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,2-叠氮乙酰氯的制备。同实施例2第一步。
第二步,2-叠氮乙酰胺基-1,6-己二酸的制备。将2-氨基-1,6-己二酸(1.61g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加2-叠氮乙酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.42g,收率61.2%。1hnmr(400mhz,d2o,ppm)δ5.83(s,2h),4.76(s,1h),2.79(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.65-2.60(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,53.4,36.1,30.1,28.1,24.2;hr-ms(c8h12n4o5):理论值:244.0808,测定值[m+h]:245.2812。
第三步,2-叠氮乙酰胺基-1,6-己二琥珀酰亚胺酯的制备。将2-叠氮乙酰胺基-1,6-己二酸(1.22g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间18-20min,合并产物流出液,40℃真空冻干24h,得到白色固体1.57g,收率71.9%。1hnmr(400mhz,dmso-d6,ppm)δ5.83(s,2h),4.76(s,1h),2.80(s,8h),2.77(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.65-2.60(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,36.1,30.1,28.1,24.2;hr-ms(c16h18n6o9):理论值:438.1135,测定值[m+h]:439.3129。
实施例9
本实施例公开了一种2-氨基-1,6-己二酸骨架、具有生物素结构的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,5-生物素酰氯的制备。同实施例3第一步。
第二步,5-生物素酰胺基-1,6-己二酸的制备。将2-氨基-1,6-己二酸(1.61g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加5-生物素酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体2.22g,收率57.5%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.77(d,j=4.8hz,2h),2.72(d,j=6.4hz,2h),2.65-2.60(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,d2o,ppm)δ179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,30.1,28.1,27.4,25.5,23.1;hr-ms(c16h25n3o6s):理论值:387.1464,测定值[m+h]:388.3589。
第三步,5-生物素酰胺基-1,6-己二琥珀酰亚胺酯的制备。将5-生物素酰胺基-1,6-己二酸(1.94g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度40-70,30min,产品出峰时间25-27min,合并产物流出液,40℃真空冻干24h,得到白色固体2.37g,收率81.5%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.81(s,8h),2.77(d,j=4.8hz,2h),2.72(d,j=6.4hz,2h),2.65-2.60(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ184.3,179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,32.4,30.1,28.1,27.4,25.5,23.1;hr-ms(c24h31n5o10s):理论值:581.1792,测定值[m+h]:582.4951。
实施例10
本实施例公开了一种2-氨基-1,7-庚二酸骨架、具有炔基富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,3-丁炔酰氯的制备。同实施例1第一步。
第二步,2-丁炔酰胺基-1,7-庚二酸的制备。将2-氨基-1,7-庚二酸(1.75g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加3-丁炔酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.66g,收率69.1%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.78(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.70(m,2h),2.65-2.60(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,76.3,71.2,53.4,36.1,35.5,30.1,28.1,24.2;hr-ms(c11h15no5):理论值:241.0950,测定值[m+h]:242.3135.
第三步,2-丁炔酰胺基-1,7-庚二琥珀酰亚胺酯的制备。将2-丁炔酰胺基-1,7-庚二酸(1.21g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间22-24min,合并产物流出液,40℃真空冻干24h,得到白色固体1.61g,收率72.2%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.80(s,8h),2.79(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.70(m,2h),2.65-2.60(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,76.3,71.2,53.4,36.1,35.5,30.3,28.1,24.2;hr-ms(c19h21n3o9):理论值:435.1278,测定值[m+h]:436.4317。
实施例11
本实施例公开了一种2-氨基-1,7-庚二酸骨架、具有叠氮富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,2-叠氮乙酰氯的制备。同实施例2第一步
第二步,2-叠氮乙酰胺基-1,7-庚二酸的制备。将2-氨基-1,7-庚二酸(1.61g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加2-叠氮乙酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.73g,收率73.5%。1hnmr(400mhz,d2o,ppm)δ5.83(s,2h),4.76(s,1h),2.79(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.70(m,2h),2.65-2.60(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,53.4,36.1,35.5,30.1,28.1,24.2;hr-ms(c9h14n4o5):理论值:258.0964,测定值[m+h]:259.3712。
第三步,2-叠氮乙酰胺基-1,7-庚二琥珀酰亚胺酯的制备。将2-叠氮乙酰胺基-1,7-庚二酸(1.29g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间18-20min,合并产物流出液,40℃真空冻干24h,得到白色固体1.21g,收率58.7%。1hnmr(400mhz,dmso-d6,ppm)δ5.83(s,2h),4.76(s,1h),2.80(s,8h),2.77(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.70(m,2h),2.65-2.60(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,36.1,32.3,30.1,28.1,24.2;hr-ms(c17h20n6o9):理论值:452.1292,测定值[m+h]:453.4265。
实施例12
本实施例公开了一种2-氨基-1,7-庚二酸骨架、具有生物素结构的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,5-生物素酰氯的制备。同实施例3第一步。
第二步,5-生物素酰胺基-1,7-庚二酸的制备。将2-氨基-1,7-庚二酸(1.75g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加5-生物素酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体2.61g,收率65.2%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.77(d,j=4.8hz,2h),2.72(d,j=6.4hz,2h),2.70(m,2h),2.65-2.60(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,d2o,ppm)δ179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,35.5,30.1,28.1,27.4,25.5,23.1;hr-ms(c17h27n3o6s):理论值:401.1621,测定值[m+h]:402.4316。
第三步,5-生物素酰胺基-1,7-庚二琥珀酰亚胺酯的制备。将5-生物素酰胺基-1,7-庚二酸(2.0g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度40-70,30min,产品出峰时间25-27min,合并产物流出液,40℃真空冻干24h,得到白色固体2.78g,收率83.2%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.81(s,8h),2.77(d,j=4.8hz,2h),2.72(d,j=6.4hz,2h),2.69(m,2h),2.65-2.60(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ184.3,179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,35.5,32.4,30.1,28.1,27.4,25.5,23.1;hr-ms(c25h33n5o10s):理论值:595.1948,测定值[m+h]:596.4691。
实施例13
本实施例公开了一种2-氨基-1,8-辛二酸骨架、具有炔基富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,3-丁炔酰氯的制备。同实施例1第一步。
第二步,2-丁炔酰胺基-1,8-辛二酸的制备。将2-氨基-1,8-辛二酸(1.89g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加3-丁炔酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.93g,收率78.2%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.78(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.70(m,2h),2.65-2.60(m,2h),2.55(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,76.3,71.2,53.4,36.1,35.5,30.1,28.1,24.2,22.9;hr-ms(c12h17no5):理论值:255.1107,测定值[m+h]:256.4127。
第三步,2-丁炔酰胺基-1,8-辛二琥珀酰亚胺酯的制备。将2-丁炔酰胺基-1,8-辛二酸(1.28g,5mmol)、edci(2.88g,15mmol)与nhs(1.89g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间22-24min,合并产物流出液,40℃真空冻干24h,得到白色固体1.96g,收率87.3%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),2.92(s,2h),2.83(s,1h),2.80(s,8h),2.79(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.70(m,2h),2.65-2.60(m,2h),2.55(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,76.3,71.2,53.4,36.1,35.5,30.3,28.1,24.2,22.1;hr-ms(c20h23n3o9):理论值:449.1434,测定值[m+h]:450.4912。
实施例14
本实施例公开了一种2-氨基-1,8-辛二酸骨架、具有叠氮富集功能的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,2-叠氮乙酰氯的制备。同实施例2第一步。
第二步,2-叠氮乙酰胺基-1,8-辛二酸的制备。将2-氨基-1,8-辛二酸(1.89g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加2-叠氮乙酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体1.56g,收率57.3%。1hnmr(400mhz,d2o,ppm)δ5.83(s,2h),4.76(s,1h),2.79(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.70(m,2h),2.65-2.60(m,2h),2.55(m,2h);13cnmr(400mhz,d2o,ppm)δ173.2,172.1,169.7,53.4,36.1,35.5,30.1,28.1,24.2,22.1;hr-ms(c10h16n4o5):理论值:272.1121,测定值[m+h]:273.3129。
第三步,2-叠氮乙酰胺基-1,8-辛二琥珀酰亚胺酯的制备。将2-叠氮乙酰胺基-1,8-辛二酸(1.36g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度30-60,30min,产品出峰时间18-20min,合并产物流出液,40℃真空冻干24h,得到白色固体1.35g,收率57.9%。1hnmr(400mhz,dmso-d6,ppm)δ5.83(s,2h),4.76(s,1h),2.80(s,8h),2.77(d,j=4.8hz,2h),2.72(d,j=4.8hz,2h),2.70(m,2h),2.65-2.60(m,2h),2.55(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ185.7,173.2,172.1,169.7,36.1,32.3,30.1,28.1,24.2,22.6;hr-ms(c18h22n6o9):理论值:466.1448,测定值[m+h]:467.5317。
实施例15
本实施例公开了一种2-氨基-1,8-辛二酸骨架、具有生物素结构的化学交联剂的制备方法,包含三个反应步骤,制备方法如下所示:
第一步,5-生物素酰氯的制备。同实施例3第一步。
第二步,5-生物素酰胺基-1,8-辛二酸的制备。将2-氨基-1,8-辛二酸(1.89g,10mmol)溶于40ml饱和碳酸氢钠溶液,降温至0℃,滴加5-生物素酰氯(12mmol)的二氯甲烷溶液,剧烈搅拌。滴毕,反应液升温至25℃,继续反应2h。反应毕,饱和氢氧化钠溶液调反应液ph为11,40ml乙酸乙酯洗水相三次,洗毕,浓盐酸调反应液ph为2,50ml乙酸乙酯萃取水相三次,合并有机相,无水硫酸钠干燥,减压蒸干有机溶剂,粗产物二氯甲烷重结晶,得到白色固体2.21g,收率53.2%。1hnmr(400mhz,d2o,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.77(d,j=4.8hz,2h),2.72(d,j=6.4hz,2h),2.70(m,2h),2.65-2.60(m,2h),2.55(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,d2o,ppm)δ179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,35.5,30.1,28.1,27.4,25.5,23.1,21.9;hr-ms(c18h29n3o6s):理论值:415.1777,测定值[m+h]:416.5612。
第三步,5-生物素酰胺基-1,8-辛二琥珀酰亚胺酯的制备。将5-生物素酰胺基-1,8-辛二酸(2.08g,5mmol)、edci(2.88g,15mmol)与nhs(1.73g,15mmol)置于50ml无水二氯甲烷中,25℃搅拌16h。反应毕,减压蒸干有机溶剂,将粗产物溶于20mldmso中,以水、乙腈为流动相,10um粒径碳十八硅球为填料,利用半制备反相色谱分离纯化,流速50ml/min,检测波长200/254nm,乙腈梯度40-70,30min,产品出峰时间26-28min,合并产物流出液,40℃真空冻干24h,得到白色固体2.27g,收率74.5%。1hnmr(400mhz,dmso-d6,ppm)δ4.76(s,1h),3.36(t,j=6.4hz,1h),3.10(s,2h),2.92(s,2h),2.86(m,4h),2.83(s,1h),2.81(s,8h),2.77(d,j=4.8hz,2h),2.72(d,j=6.4hz,2h),2.69(m,2h),2.65-2.60(m,2h),2.55(m,2h),2.05(t,j=6.4hz,2h),1.57(m,2h),1.25(m,2h);13cnmr(400mhz,dmso-d6,ppm)δ184.3,179.4,173.2,172.1,164.7,76.3,63.3,62.4,55.6,53.4,38.2,36.7,35.5,32.4,30.1,28.1,27.4,25.5,23.1,21.9;hr-ms(c26h35n5o10s):理论值:609.2105,测定值[m+h]:610.6418。
实施例16
本实施例公开了化学交联剂对jurkat细胞质膜蛋白质进行相互作用信息鉴定,其中化学交联剂选择实施例1中以天冬氨酸骨架、具有炔基富集功能的化学交联剂,具体过程如下:
细胞的获取:将培养的jurkat细胞用4℃预冷的1х磷酸盐缓冲液洗3次,离心3000rpm,5min,4℃。
化学交联反应:将所得细胞计数,1*108细胞加入1ml1х磷酸盐缓冲液(ph7.4)+1%天冬氨酸炔基交联剂(使用dmso溶解),交联剂初始浓度为20mm,dmso:水相=1:99,室温,反应1h。3000rpm离心,5min,4℃,去掉上清液。
还原、烷基化:加入相同体积的终浓度为100mm的碳酸氢铵终止交联,同时其中含有25mm三(2-羧乙基)膦盐酸盐,将细胞打散后,再加入终浓度为20mm碘代乙酰胺,室温避光反应30min。离心3000rpm,5min,4℃,去掉上清液。
酶解:加入相同体积的1mg/ml的蛋白酶k,37℃酶解1h。
获得肽段样品:40000g离心,30min,4℃。取上清,即为肽段样品。
将上述得到的肽段样品进行质谱鉴定,使用orbitrapfusionlumos质谱仪,hcd的质谱采集模式。
将得到的质谱数据使用plink2软件进行检索,共鉴定到152对cross-link型交联肽段,经进一步分析,其中132对为质膜蛋白质的相互作用肽段,占总交联肽比例的86.8%。
上述数据结果表明,通过使用天冬氨酸炔基交联剂作为化学交联剂对jurkat细胞进行化学交联反应,共鉴定到198对相互作用肽段,其中92.5%为质膜蛋白质相互作用肽段,实现了对人源细胞jurkat细胞质膜蛋白质相互作用信息的高效规模化鉴定。
- 还没有人留言评论。精彩留言会获得点赞!