一种聚醚型多硫醇化合物的制备方法与流程
本发明涉及有机合成
技术领域:
,特别涉及一种聚醚型多硫醇化合物的制备方法。
背景技术:
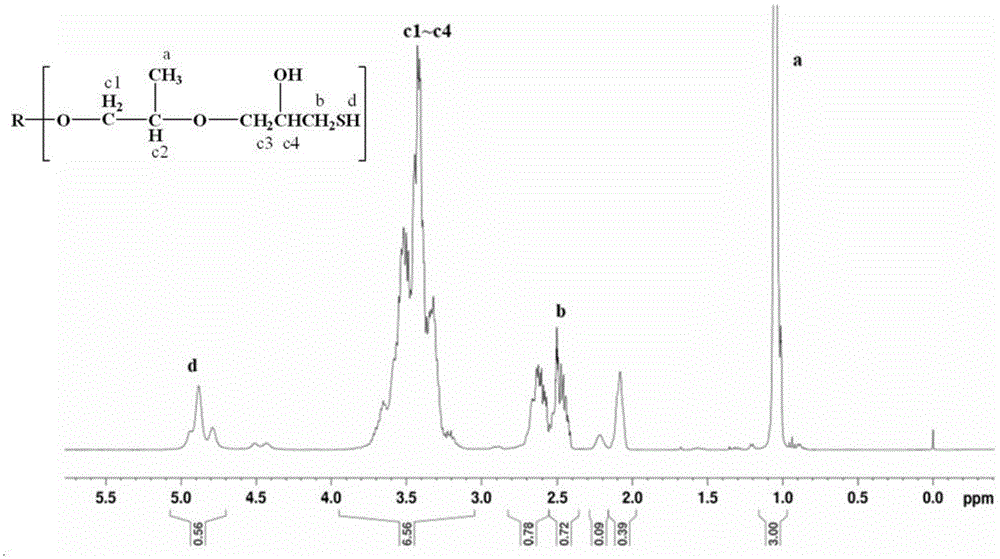
:环氧树脂是合成树脂的一个重要品种,它具有粘结性强,收缩率小,机械性能好,介电性能和耐热性能优良等优点,可作为涂料、胶黏剂等广泛应用于机械、电子、建筑等领域。但其作为固化剂使用时,由于其低温固化不完全、固化速度慢等缺陷,而限制了其在固化剂领域的应用。而聚硫醇在叔胺的促进下显示出优异的低温快速固化性能,成为低温条件下的最佳固化剂。具体的,聚硫醇固化剂末端为硫醇基,在室温下单独使用时反应缓慢、几乎不能进行,但在适当的促进剂下,聚硫醇会形成硫醇离子,固化反应速度是多元胺固化剂的数倍,这个特点在低温固化时更加明显。常温下,传统聚酰胺环氧固化剂需要24小时才能固化完全,而聚硫醇体系只需5分钟,因此,在室温和低温条件下,聚硫醇类固化剂是无法替代的。另一方面,硫醇类固化剂因为含有硫柔性链段并具有高折射率,在透明树脂和增韧树脂方面也有很大的应用价值。而目前国内生产极少,一般为进口美国capcure系列产品或日本epomate系列产品。因此,聚硫醇固化剂的开发和制备具有重要意义。廖毅彬等公开了一种聚硫醇固化剂的制备方法(聚硫醇固化剂的制备及其活性研究,厦门大学理学硕士学位论文,2014,4),采用聚醚多元醇ge303和环氧氯丙烷进行开环反应制得含有醚氯醇的中间体pce,再将中间体pce与硫氢化钠溶液进行亲核取代反应,得到聚醚型聚硫醇。然而,该方法还存在明显的缺陷:其必须采用特定有机溶剂工业乙醇才能保证反应顺利进行,并获得高巯基含量产品。但是有机溶剂成本高,使工业生产成本大幅提升,且有机溶剂易散失到环境中造成污染。而以水为溶剂时,虽克服了上述成本和环境污染的问题,但是反应过程难以顺利进行,氯醇还易发生环化反应,难以获得高收率和转化率、及高巯基含量的聚硫醇产品。另外,无论以水为溶剂还是有机溶剂为溶剂,所得产品都为黄绿色或浅黄色产品,透明度差,无法用于透明度要求较高如珠宝等高端产品的粘合,限制了其应用范围;且所得产品氯离子含量高,不适用于儿童玩具、电子元件等的制作,也大大局限了其应用。技术实现要素:有鉴于此,本发明的目的在于提供一种聚醚型多硫醇化合物的制备方法,本发明的制备方法,以水为溶剂仍能保证反应顺利进行,并获得高转化率和收率,以及高巯基含量产品;另外,本发明的制备方法所得的聚硫醇产品为无色透明,氯离子含量低,有利于扩展其应用范围。本发明提供了一种聚醚型多硫醇化合物的制备方法,包括以下步骤:a)在路易斯酸催化剂的作用下,将式(1)所示聚醚多元醇与环氧氯丙烷反应,得到式(2)所示氯代聚醚;所述环氧氯丙烷与聚醚多元醇的摩尔比为2.0~10.0;b)在相转移催化剂的作用下和h2s气体环境下,将所述氯代聚醚与nahs水溶液进行取代反应,得到式(3)所示聚醚型多硫醇化合物;所述相转移催化剂选自季铵盐类化合物和氮杂环类化合物中的一种或几种;优选的,所述相转移催化剂选自苄基三乙基氯化铵、四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵、三辛基甲基氯化铵、十二烷基三甲基氯化铵、十四烷基三甲基氯化铵、吡啶和三丁胺中的一种或几种。优选的,所述相转移催化剂选自苄基三乙基氯化铵、四丁基溴化铵、四丁基氯化铵和四丁基硫酸氢铵中的一种或几种;所述相转移催化剂的用量为所述氯代聚醚的1wt%~20wt%。优选的,所述nahs水溶液的浓度为10wt%~50wt%;所述nahs与环氧氯丙烷的摩尔比为0.5~3.0。优选的,所述步骤b)中,取代反应的温度为70~180℃,时间为2~100小时。优选的,所述步骤b)中,取代反应的温度为80~120℃,时间为5~20小时。优选的,所述路易斯酸催化剂选自bf3、alcl3、sbcl5、sncl4、ticl4、febr3、fecl3和zncl2中的一种或几种;所述路易斯酸催化剂的用量为所述聚醚多元醇的0.1wt%~10wt%。优选的,所述步骤a)中,反应的温度为40~150℃,时间为2~100小时。优选的,在所述取代反应后,还包括后处理;所述后处理包括:c)对所述取代反应所得粗产品进行一次水洗后,调节ph值至3~9,然后进行二次水洗,得到水洗产物;d)对所述水洗产物进行真空除水,得到聚醚型多硫醇化合物。优选的,所述真空除水的真空度≤-0.095mpa,温度为60~80℃,时间为1~3小时。本发明提供了一种聚醚型多硫醇化合物的制备方法,先将聚醚多元醇与环氧氯丙烷反应,得到中间产物氯代聚醚,再将所得中间产物氯代聚醚与nahs水溶液进行取代反应,得到聚醚型多硫醇化合物;本发明在上述制备过程中,采用nahs水溶液即以水为溶剂进行合成反应,控制第二步反应采用在特定相转移催化剂及特定的h2s气氛下进行,结果能够顺利制备聚醚型多硫醇化合物,并获得高转化率和收率,以及高巯基含量产品;另外,本发明的制备方法所得的聚硫醇产品为无色透明,氯离子含量低,有利于扩展其应用范围。试验结果表明,本发明的制备方法的转化率在99%以上,收率在82%以上,产品巯基含量在11%以上,色度在30以下,氯含量在800ppm以下。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。图1为本发明实施例1所得产物的氢核磁1hnmr测试图。具体实施方式本发明提供了一种聚醚型多硫醇化合物的制备方法,包括以下步骤:a)在路易斯酸催化剂的作用下,将式(1)所示聚醚多元醇与环氧氯丙烷反应,得到式(2)所示氯代聚醚;所述环氧氯丙烷与聚醚多元醇的摩尔比为2~10;b)在相转移催化剂的作用下和h2s气体环境下,将所述氯代聚醚与nahs水溶液进行取代反应,得到式(3)所示聚醚型多硫醇化合物;所述相转移催化剂选自季铵盐类化合物和氮杂环类化合物中的一种或几种;本发明先将聚醚多元醇与环氧氯丙烷反应,得到中间产物氯代聚醚,再将所得中间产物氯代聚醚与nahs水溶液进行取代反应,得到聚醚型多硫醇化合物;本发明在上述制备过程中,采用nahs水溶液即以水为溶剂进行合成反应,控制第二步反应采用在特定相转移催化剂及特定的h2s气氛下进行,结果能够顺利制备聚醚型多硫醇化合物,且其收率和转化率高,所得产品具有高巯基含量,克服了现有制备方法中以水为溶剂时的缺陷;另外,所得产品为无色透明,且氯含量较低,克服了现有制备方法中产品色度差、氯含量高的问题。按照本发明,先在路易斯酸催化剂的作用下,将式(1)所示聚醚多元醇与环氧氯丙烷反应,得到式(2)所示氯代聚醚;本发明中,所述路易斯酸催化剂优选为bf3、alcl3、sbcl5、sncl4、ticl4、febr3、fecl3和zncl2中的一种或几种;更优选为alcl3、sbcl5、sncl4和ticl4中的一种或几种。本发明对所述路易斯酸催化剂的来源没有特殊限制,为一般市售品即可。作为优选,所述路易斯酸催化剂的用量为所述聚醚多元醇的0.1wt%~10wt%,更优选为0.3wt%~5wt%。本发明中,所述式(1)所示聚醚多元醇中,r没有特殊限制,为本领域技术人员熟知的聚醚多元醇种类即可。式(1)中,n优选为2~200之间的整数,更优选为5~50之间的整数。本发明对所述式(1)所示聚醚多元醇的来源没有特殊限制,为一般市售品或按照本领域技术人员熟知的制备方式获得即可。本发明中,所述环氧氯丙烷与聚醚多元醇的摩尔比为2.0~10.0,优选为2.5~6.0。本发明中,式(1)所示聚醚多元醇与环氧氯丙烷反应的温度优选为40~150℃,更优选为60~100℃;在本发明的一些实施例中,反应的温度为90~100℃。本发明中,所述反应的时间优选为1~100小时,更优选为5~20小时;在本发明的一些实施例中,所述反应的时间为7~10小时。在本发明的一些实施例中,反应的温度为100℃,时间为7小时。本发明中,作为优选,所述步骤a)具体包括:a1)将路易斯酸催化剂与式(1)所示聚醚多元醇混合溶解,得到溶解物;a2)将所述溶解物与环氧氯丙烷混合反应,得到式(2)所示氯代聚醚。其中,所述步骤a1)中,溶解的温度优选为60~75℃。所述步骤a2)中,优选以滴加的方式引入环氧氯丙烷。本发明中,式(1)所示聚醚多元醇与环氧氯丙烷反应生成式(2)所示氯代聚醚,式(2)中,n优选为2~200之间的整数,更优选为5~50之间的整数。按照本发明,在得到式(2)所示氯代聚醚后,在相转移催化剂的作用下和h2s气体环境下,将所述氯代聚醚与nahs水溶液进行取代反应,得到式(3)所示聚醚型多硫醇化合物。本发明中,所述相转移催化剂为季铵盐类化合物和氮杂环类化合物中的一种或几种;优选为苄基三乙基氯化铵、四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵、三辛基甲基氯化铵、十二烷基三甲基氯化铵、十四烷基三甲基氯化铵、吡啶和三丁胺中的一种或几种;更优选为苄基三乙基氯化铵、四丁基溴化铵、四丁基氯化铵和四丁基硫酸氢铵中的一种或几种。本发明在第二步水溶剂反应体系中引入上述相转移催化剂,有利于促进体系巯基化反应的进行,提高转化率及收率。作为优选,所述相转移催化剂的用量为所述氯代聚醚的1wt%~20wt%,更优选为5wt%~12wt%。本发明中,第二步取代反应在h2s气体环境下进行。本发明的水溶剂体系中,采用h2s气体环境与相转移催化剂配合,二者协同,能够有效抑制副反应的发生,有利于获得高转化率和收率。本发明对所述h2s气体的气压没有特殊限制,为常压即可。本发明中,所述nahs水溶液的浓度优选为10wt%~50wt%,更优选为15wt%~44wt%;在上述浓度范围内,有利于保证反应顺利进行,既不会造成反应剧烈,导致反应不均、产物颜色较深,也不会造成反应难以进行,导致转化率和收率较低。本发明中,所述nahs水溶液的浓度是指在整个反应体系内,nahs在水中的浓度。所述nahs水溶液的引入形式没有特殊限制,可将上述目标浓度的nahs水溶液直接投入反应体系;也可先投入一定浓度的nahs水溶液,再向反应体系内加水调节使反应体系内nahs在水中的浓度达到上述目标浓度。优选的,先加入一定浓度的nahs水溶液(记为初级nahs水溶液),再向反应体系内加入水调节初级nahs水溶液的浓度,保证整个反应体系内,nahs水溶液的浓度达至上述目标浓度。本发明中,所述nahs与环氧氯丙烷的摩尔比优选为0.5~3.0,更优选为0.9~1.5。本发明中,所述取代反应的温度优选为70~180℃,更优选为80~120℃;在本发明的一些实施例中,所述取代反应的温度为85℃或90℃。所述取代反应的时间优选为2~100小时,更优选为5~20小时。现有技术的醇溶剂体系制备时,nahs亲核取代反应的温度须严格控制在65℃以下,才能获得聚醚多硫醇产物;而本发明的水溶剂体系中,取代反应的温度须控制在70℃以上才能保证反应顺利进行并获得高转化率和产率,以及无色透明产品。本发明中,所得式(3)所示聚醚型多硫醇化合物中,n优选为2~200之间的整数,更优选为5~50之间的整数。本发明在相转移催化剂的作用下和h2s气体环境下,将氯代聚醚与nahs水溶液进行取代反应,成功得到式(3)所示聚醚型多硫醇化合物。现有技术中须在特定有机醇溶剂(工业乙醇)中,才能促进硫氢化钠溶解并解离为sh-,促进亲核反应进行,获得高巯基含量产品;采用水溶剂时,反应过程难以顺利进行,氯醇还易发生环化反应,难以获得高收率和转化率、及高巯基含量的聚硫醇产品。而本发明采用水溶剂体系控制第二步反应采用在特定相转移催化剂及特定的h2s气氛下进行,以及配合反应条件,结果能够顺利制备聚醚型多硫醇化合物,且其收率和转化率高,所得产品具有高巯基含量,克服了现有制备方法中以水为溶剂时的缺陷;另外,所得产品为无色透明,且氯含量较低,克服了现有制备方法中产品色度差、氯含量高的问题,扩展了聚醚型多硫醇化合物固化剂的应用范围。另外,现有技术需采用高纯nahs固体,而本发明采用工业级nahs液体,大大降低了成本。本发明中,所述步骤b)的取代反应优选在釜式反应器中进行。现有技术中,采用的是加热回流法,需要在回流装置中进行,过程中须严格控制中间产物的氯代聚醚的滴加速度,操作繁琐复杂,费时费力,难以实现工业化生产;而本发明在常规反应釜中进行取代反应,像常规聚合反应一样将物料一次性投入即可,无需监控滴加,相比于现有技术,大大简化了操作,便于工业化生产。另外,现有技术采用的是回流装置,需先将醇溶剂和nahs固体在回流装置中溶解后再进行后续反应,而本发明采用反应釜,直接将nahs水溶液投料即可,操作上也更为简单。本发明中,在进行取代反应后,优选还包括后处理;所述后处理优选包括:c)对所述取代反应所得粗产品进行一次水洗后,调节ph值至3~9,然后进行二次水洗,得到水洗产物;d)对所述水洗产物进行真空除水,得到聚醚型多硫醇化合物。在所述取代反应后,优选先静置分层,上层液体即为粗产品a。得到粗产品a后,进行一次水洗。具体的,向粗产品a中加入一定量的水;加水量优选为粗产品a质量的1~2倍。本发明中,所述一次水洗的温度优选为60~100℃。之后,调节ph至3~9,优选调节ph至5~7。优选加酸调节ph;本发明对所述酸没有特殊限制,为本领域技术人员熟知的用于调节ph至酸性的试剂即可,如硫酸或盐酸等。调节ph后,优选再进行静置分层,取出下层物质即为粗产品b。然后进行二次水洗,加水量优选与一次加水的加水量相同。本发明中,所述二次水洗的温度优选为60~100℃。二次加水后,优选再进行静置处理,静置后分层,取出下层物质,即为水洗产物。得到水洗产物后,进行真空除水处理。本发明中,所述真空除水的真空度≤-0.095mpa,温度为60~70℃,时间为1~3小时。在本发明的一些实施例中,真空除水的温度为65℃,时间为2小时。在上述真空除水处理后,即得式(3)聚醚型多硫醇化合物。本发明提供了一种聚醚型多硫醇化合物的制备方法,先将聚醚多元醇与环氧氯丙烷反应,得到中间产物氯代聚醚,再将所得中间产物氯代聚醚与nahs水溶液进行取代反应,得到聚醚型多硫醇化合物。本发明无需使用有机溶剂,在水溶剂体系中仍能成功制备聚醚多硫醇,且获得高转化率和收率,以及高巯基含量,另外,所得产品无色透明,氯离子含量低;且本发明的方法操作简单,便于工业化生产。所得聚醚多硫醇产品与环氧树脂相溶性好、固化速度快,由于具有柔性结构固化后剪切应力强,可在低温及室温下直接操作无需加热,且性质稳定,长期储存后不会有沉淀和结晶现象。为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。以下实施例中,各原料及试剂均为市售商品,其中,聚醚多元醇型号为yd-303,由河北亚东化工集团提供。实施例1取89.6g聚醚多元醇和0.46g路易斯酸催化剂bf3,升温至65℃使催化剂全部溶解后,滴加87.9g环氧氯丙烷(环氧氯丙烷与聚醚多元醇的摩尔比为3.90),滴加过程中控制反应温度在100℃,反应时间为7小时,反应完成后,得到氯代聚醚。向反应釜内加入198.3g质量分数为32%的nash水溶液(nash与环氧氯丙烷的摩尔比为1.2)和13.3g相转移催化剂四丁基硫酸氢铵,然后加入177.7g氯代聚醚和132g水,将反应釜封闭后检查气密性,置换后向釜内通入h2s气体使釜内处于常压,通过油浴加热反应体系至90℃后保温反应6小时,然后开启反应釜的放空阀并用n2置换,产物静置分层后,得到粗产品a;将粗产品a加入180g去离子水中进行一次水洗,用盐酸调节有机相ph至5~7,待稳定后静置分层,取出下层物质即为粗产品b;向粗产品b中加入180g去离子水混合均匀后静置12小时,然后切出产物并在真空度≤-0.095mpa条件下进行除水,除水温度为65℃,时间为2小时,得到无色透明聚醚型多硫醇产物155.2g。对所得产物进行氢核磁1hnmr检测,结果如图1所示,图1为本发明实施例1所得产物的氢核磁1hnmr测试图,可以看出,所得产品为式(3)所示聚醚型多硫醇化合物。计算产品的转化率和收率,结果参见表1。实施例2取85.2g聚醚多元醇和0.46g路易斯酸催化剂zncl2,升温至65℃使催化剂全部溶解后,滴加70.3g环氧氯丙烷(环氧氯丙烷与聚醚多元醇的摩尔比为3.3),滴加过程中控制反应温度在100℃,反应时间为7小时,反应完成后,得到氯代聚醚。向反应釜内加入175.7g质量分数为32%的nash水溶液(nash与环氧氯丙烷的摩尔比为1.2)和8.6g相转移催化剂四丁基硫酸氢铵,然后加入172.0g氯代聚醚和127.8g水,将反应釜封闭后检查气密性,置换后向釜内通入h2s气体使釜内处于常压,通过油浴加热反应体系至85℃后保温反应6小时,然后开启反应釜的放空阀并用n2置换,产物静置分层后,得到粗产品a;将粗产品a加入173g去离子水中进行一次水洗,用盐酸调节有机相ph至5~7,待稳定后静置分层,取出下层物质即为粗产品b;向粗产品b中加入173g去离子水混合均匀后静置10小时,然后切出产物并在真空度≤-0.095mpa条件下进行除水,除水温度为65℃,时间为2小时,得到无色透明聚醚型多硫醇产物151.1g。按照实施例1的检测方法对所得产物进行检测,结果显示,其为式(3)所示聚醚型多硫醇化合物。计算产品的转化率和收率,结果参见表1。实施例3取96.1g聚醚多元醇和0.52g路易斯酸催化剂sncl4,升温至65℃使催化剂全部溶解后,滴加86.9g环氧氯丙烷(环氧氯丙烷与聚醚多元醇的摩尔比为3.6),滴加过程中控制反应温度在100℃,反应时间为7小时,反应完成后,得到氯代聚醚。向反应釜内加入125.3g质量分数为32%的nash水溶液(nash与环氧氯丙烷的摩尔比为1.2)和8.1g相转移催化剂四丁基硫酸氢铵,然后加入161.0g氯代聚醚和95.3g水,将反应釜封闭后检查气密性,置换后向釜内通入h2s气体使釜内处于常压,通过油浴加热反应体系至85℃后保温反应6小时,然后开启反应釜的放空阀并用n2置换,产物静置分层后,得到粗产品a;将粗产品a加入175g去离子水中进行一次水洗,用盐酸调节有机相ph至5~7,待稳定后静置分层,取出下层物质即为粗产品b;向粗产品b中加入175g去离子水混合均匀后静置10小时,然后切出产物并在真空度≤-0.095mpa条件下进行除水,除水温度为65℃,时间为2小时,得到无色透明聚醚型多硫醇产物154.2g。按照实施例1的检测方法对所得产物进行检测,结果显示,其为式(3)所示聚醚型多硫醇化合物。计算产品的转化率和收率,结果参见表1。实施例4取91.6g聚醚多元醇和0.49g路易斯酸催化剂bf3,升温至65℃使催化剂全部溶解后,滴加86.3g环氧氯丙烷(环氧氯丙烷与聚醚多元醇的摩尔比为3.9),滴加过程中控制反应温度在100℃,反应时间为7小时,反应完成后,得到氯代聚醚。向反应釜内加入135.2g质量分数为32%的nash水溶液(nash与环氧氯丙烷的摩尔比为1.2)和8.5g相转移催化剂四丁基硫酸氢铵,然后加入170.0g氯代聚醚和102.7g水,将反应釜封闭后检查气密性,置换后向釜内通入h2s气体使釜内处于常压,通过油浴加热反应体系至85℃后保温反应6小时,然后开启反应釜的放空阀并用n2置换,产物静置分层后,得到粗产品a;将粗产品a加入180g去离子水中进行一次水洗,用盐酸调节有机相ph至3~4,待稳定后静置分层,取出下层物质即为粗产品b;向粗产品b中加入180g去离子水混合均匀后静置10小时,然后切出产物并在真空度≤-0.095mpa条件下进行除水,除水温度为65℃,时间为2小时,得到无色透明聚醚型多硫醇产物156.8g。按照实施例1的检测方法对所得产物进行检测,结果显示,其为式(3)所示聚醚型多硫醇化合物。计算产品的转化率和收率,结果参见表1。实施例5~7实施例5:按照实施例1的制备过程进行,不同的是,相转移催化剂替换为四丁基氯化铵。实施例6:按照实施例2的制备过程进行,不同的是,相转移催化剂替换为四丁基溴化铵。实施例7:按照实施例3的制备过程进行,不同的是,相转移催化剂替换为苄基三乙基氯化铵。按照实施例1的检测方法对实施例5~7所得产物进行检测,结果显示,所得产物为式(3)所示聚醚型多硫醇化合物。计算产品的转化率和收率,结果参见表1。实施例8实施例1~7所得产品的各项检测结果参见表1,各项目的检测依据如下:①巯基含量(-sh含量,%):通过滴定法测试,具体原理是:试样被溶剂溶解后用碘将硫醇氧化为二硫化物,硫代硫酸钠标准溶液滴定剩余的碘,实现巯基的定量;②氯含量(ppm):通过库仑氯滴定测试,其原理是:将试样注入燃烧管,与氧气混合并燃烧,试样中的有机氯转化为氯化氢,并由载气带入滴定池,与电解液中的银离子发生反应(ag++cl-→agcl↓),消耗的银离子由微库仑计通过电解补充,根据反映所需要的电量,按照法拉第电解定律计算试验中的氯含量;③色度(hazen):通过亨特立色度仪测试;④粘度(pa.s,25℃):通过ndj-5s数显粘度计测试;⑤凝胶时间:按照国标gb/t12007.7-1989对固化体系进行凝胶时间测试。表1实施例1~7的检测结果转化率收率-sh含量氯含量色度粘度(25℃)凝胶时间实施例199.2%89.1%12.9%561ppm2012.9pa.s2min实施例299.3%85.6%11.3%620ppm2611.5pa.s4min实施例399.1%82.9%11.9%781ppm2810.9pa.s3min实施例499.2%84.5%12.3%673ppm219.5pa.s3min实施例599.8%87.6%12.4%508ppm2311.2pa.s4min实施例699.4%88.1%11.9%496ppm2913.0pa.s3min实施例799.1%86.2%13.0%801ppm3010.6pa.s3min由以上测试结果可知,采用本发明的方法简单便捷,无需使用有机溶剂,且其选择性好、原子利用率高,能够获得高转化率和收率,且产品巯基含量在11%以上,色度在30以下,粘度高,氯含量低,凝胶时间快。对比例1按照现有技术“聚硫醇固化剂的制备及其活性研究”公开的聚醚型多硫醇的制备方法制备,不同的是,制备过程采用水为溶剂。具体过程如下:s1:取96.1g聚醚多元醇(ge303)、结晶四氯化锡催化剂(为聚醚多元醇ge303用量的0.5wt%),依次加入装有冷凝管、搅拌器、恒压漏斗及加热器的250ml三颈瓶中。通冷凝水,开启搅拌器和加热器,在70℃左右开始滴加环氧氯丙烷(环氧氯丙烷与聚醚多元醇中羟基的摩尔比为1.2∶1),控制环氧氯丙烷的滴加速度,使反应温度不超过80℃。滴加完毕,在80℃回流反应1.5h,自然冷却,得到中间产物pce。s2:称取硫氢化钠40.1g和水266.8g,依次加入装有冷凝管、搅拌器、分液漏斗及加热器的250ml三颈瓶中。通冷凝水,开启搅拌器和加热器,在55℃左右开始滴加pce,控制pce的滴加速度,使反应温度不超过65℃。滴加完毕,在65℃回流反应2h,将产物倒入分液漏斗中,自然冷却。将上层黄绿色有机物,用体积比为1∶1的稀盐酸中和至ph为5~6。s3:用抽滤装置提取母液,用减压蒸馏装置,开启真空泵及加热器进行减压蒸馏,至无液体抽出为止,得到棕褐色混浊液体,于分液漏斗中分层得油状物,真空烘干,得到黄绿色聚硫醇产物。对比例2按照对比例1的制备过程进行,不同的是,将溶剂水替换为工业乙醇;所得产物为淡黄色。按照实施例8的测试方法对对比例1~2进行检测,结果参见表2。表2对比例1~2的检测结果转化率收率-sh含量氯含量色度粘度(25℃)凝胶时间对比例198.0%80.0%9.8%1030ppm7911.7pa.s5min对比例298.3%81.7%10.3%1164ppm6310.9pa.s5min由以上测试结果可知,按照现有技术的制备方法,以水为溶剂时,反应转化率和收率较低,巯基含量也较低,氯含量较高,色度较差;以工业乙醇为溶剂时,产品巯基含量较低,氯含量较高,色度较差,且使生产成本大大提升。相比之下,本发明的制备方法,转化率和收率较高,且产品巯基含量高,同时,产品无色透明、具有较好的色度,且氯含量较低,克服了以上问题。同时,根据对比例1~2可知,其制备过程复杂,条件控制要求高;相比之下,本发明的制备方法简单易行,更有利于实现规模化生产。以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1