从朝鲜蓟中提取愈创木烷型倍半萜的方法与流程
本发明涉及提取纯化领域,具体而言,涉及一种从朝鲜蓟中提取愈创木烷型倍半萜的方法。
背景技术:
朝鲜蓟为菊科菜蓟属多年生草本植物,又名菜蓟、洋蓟、菊蓟、荷花百合、法国百合,学名cynarascolymusl.。原产于地中海沿岸,欧洲南部及北非,19世纪由法国传入中国上海,现主要分布在上海、浙江、云南、湖南、山东及北京等地。朝鲜蓟食用价值极高,有“蔬菜之皇”的美誉。
朝鲜蓟叶提取物长期以来一直用于民间医药,其体外和临床试验表明,朝鲜蓟提取物具有抗氧化、抑菌、抗癌抗肿瘤、保肝、降血糖、改善消化及改善高胆固醇血症等功效,愈创木烷半倍萜内酯作为其主要功效成分有望被开发为治疗炎症性疾病(如风湿性关节炎)、特异性抗乙型肝炎病毒、ii型糖尿病相关的新型ptp-1b抑制剂、血管生成抑制剂和治疗多种癌症的新药物以及促进胃动力和治疗胃溃疡的新药物。
但是现有技术对于其有效成分的分离纯化研究有限。如何从朝鲜蓟中提取出愈创木烷型倍半萜化合物等生物活性成分,对深度开发朝鲜蓟作物极其重要。
有鉴于此,特提出本发明。
技术实现要素:
本发明的目的在于提供一种从朝鲜蓟中提取愈创木烷型倍半萜的方法,从朝鲜蓟中提取出目标化合物,而且快速有效、纯度高。
为了实现本发明的上述目的,特采用以下技术方案:
一种从朝鲜蓟中提取愈创木烷型倍半萜的方法,所述方法包括以下步骤:
a.将朝鲜蓟粉碎后用乙醇水溶液浸提后得到粗提液;
b.所述粗提液上样至ab-8型大孔树脂层析柱,依次用水、体积分数为20%、50%、80%的乙醇水溶液进行洗脱,经色谱检测,收集含有目标化合物的洗脱液,浓缩冻干得到粗提物;
c.所述粗提物用体积分数为50%的甲醇水溶液溶解后上样至rp-c18层析柱,依次用体积分数为30%、48%和100%的甲醇水溶液进行洗脱,经色谱检测,收集含有所述目标化合物的洗脱液,浓缩冻干得到预分离物;
d.将所述预分离物用体积分数为30%的甲醇水溶液溶解,使用venusilmpc18色谱柱、以流动相a为甲醇、流动相b为甲酸水溶液作为流动相进行液相半制备,收集含有目标化合物的流分段,浓缩冻干得到目标化合物,所述目标化合物的结构式为:
使用浸提-ab-8型大孔树脂柱层析-rp-c18柱层析-甲醇/甲酸水溶液液相半制备的方法,有效的将目标化合物从朝鲜蓟中分离出来。
优选地,所述ab-8型大孔树脂层析柱的制作方法为:先将ab-8型大孔树脂颗粒用无水乙醇浸泡20-24h进行预处理,然后装柱,用水冲洗至无醇味。
预处理是为了有效的保障大孔树脂柱层析的分离效果,避免杂质的干扰。
优选地,所述rp-c18层析柱的制备方法为:将rp-c18颗粒放入甲醇中进行超声处理,并不断搅拌,去除气泡;然后将处理好的所述rp-c18颗粒和甲醇一同装柱。
更加优选地,所述rp-c18颗粒的规格为:40-60μm,
c18层析柱填料和制备方法的选择,也是为了将目标化合物从复杂的成分中最大程度的有效分离出来。用甲醇超声处理填料可以有效的去除填料的杂质,去除气泡,保证柱层析的效果。
优选地,所述venusilmpc18色谱柱的规格为:10mm×250mm,5μm。
进一步优选地,所述流动相b中甲酸水溶液的体积分数为0.1%。
更加优选地,所述液相半制备的洗脱梯度为:初始-30分钟,所述流动相a与所述流动相b的体积比为30:70;31分钟-40分钟,所述流动相a与所述流动相b的体积比为100:0。
合适的色谱柱、流动相和洗脱梯度,能够快速、高效的将复杂成分中的目标化合物进一步分离出来,使得分离变得高效可控。
优选地,所述步骤a中:所述朝鲜蓟与所述乙醇水溶液的料液比为1:8-10。
进一步优选地,浸提时间为70-72h。
可选地,所述乙醇水溶液的体积分数为70%。
浸提方法的优化,一方面是为了提高提取得率,另一方面要保证有一定的选择性,同时还不能破坏目标化合物的结构。
与现有技术相比,本发明的有益效果为:
(1)从朝鲜蓟中分离出了目标化合物;
(2)本申请提供的提取方法简单、有效,适合于推广应用。
(3)目标化合物提取纯度高。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,以下将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
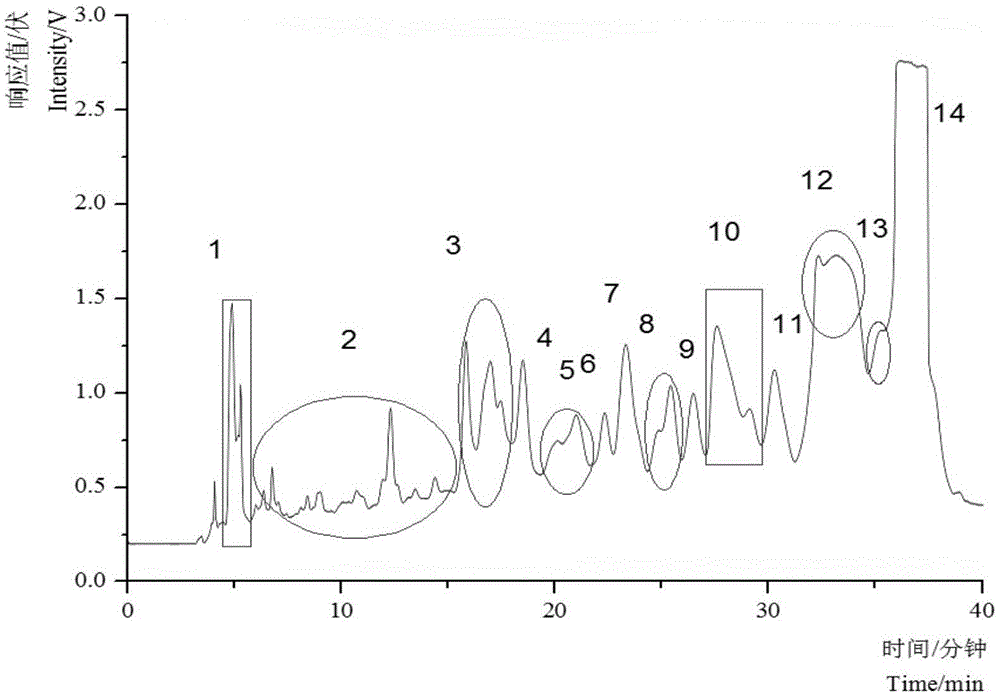
图1为实施例1进行液相半制备的谱图;
图2为实施例1制备得到的目标化合物的纯度测定图;
图3为本申请制备得到的目标化合物的质谱图;
图4为本申请制备得到的目标化合物的h谱图;
图5为本申请制备得到的目标化合物的c谱图;
图6为本申请制备得到的目标化合物的h-hcosy谱图;
图7为本申请制备得到的目标化合物的hsqc谱图;
图8为本申请制备得到的目标化合物的hmbc谱图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
实施例1
将朝鲜蓟晒干并用粉碎机进行粉碎,在室温条件下,将朝鲜蓟粉末按料液比1:10加入体积分数为70%的乙醇水溶液中浸提72小时,浸提期间要不断搅拌,然后提取液先用滤布真空抽滤,再用滤纸真空抽滤,将滤液减压浓缩得粗提液。粗提液的含固量为0.1g/ml。
将ab-8型大孔树脂用无水乙醇浸泡24h,浸泡期间须不断搅拌。之后,将树脂颗粒和无水乙醇一同装柱(柱体积为1l),并用无水乙醇洗至流出液加水(1:5)不浑浊,再用纯水冲洗至无醇味。将柱子内的纯水放至与树脂相平时,将1l粗提液缓慢加入到层析柱中,到柱子下层的树脂全部被染上颜色后开始接收有明显颜色的流出液(浅绿色、棕黄色),进液相分析,以免样品过多,大孔树脂吸附不完全,出现过载现象。待样品被完全吸附后,依次用水、体积分数为20%的乙醇水溶液、体积分数为50%的乙醇水溶液和体积分数为80%的乙醇水溶液进行洗脱,洗脱至无色(一直不变),同时用干净玻璃瓶接出流分(每400ml接一个流分)。将接出的流分编号为a1、a2、a3……。将收集的流分过0.45μm滤膜过滤,然后进行高效液相色谱检测。液相色谱检测条件:色谱柱型号为venusilmpc18,4.6mm×250mm,5μm;检测波长为205nm,流动相a为甲醇,流动相b为0.1%甲酸水,进样量20μl。洗脱梯度为:初始,流动相a与流动相b的体积比为5:95;至30分钟变化为100%甲醇。根据检测结果进行合并,选择活性组分a27-29进一步分离和纯化,浓缩冻干得到粗提物。
将rp-c18填料(60μm,
将预分离物用体积分数为30%的甲醇水溶液溶解,使用lc3000型高效液相色谱仪、venusilmpc18色谱柱(10mm×250mm,5μm)、以流动相a为甲醇、流动相b为体积分数0.1%的甲酸水溶液作为流动相,洗脱梯度为:初始-30分钟,流动相a与流动相b的体积比为30:70;31分钟-40分钟,流动相a与流动相b的体积比为100:0。控制进样量80μl、流速3ml/min、检测波长205nm,进行液相半制备,收集含有目标化合物的流分段(见图1,12号峰),浓缩冻干得到目标化合物。
对目标化合物进行纯度测定:以甲醇-甲酸水溶液(甲醇与甲酸水溶液的体积比为30:70,甲酸水溶液的体积分数为0.1%)作为流动相,用lc3000型高效液相色谱仪,venusilmpc18(4.6mm×250mm,5μm),检测器:uv205nm,控制流速1ml/min。测得纯度为96.1%(见图2),提取得率为109.1mg/kg。
实施例2
将朝鲜蓟晒干并用粉碎机进行粉碎,在室温条件下,将朝鲜蓟粉末按料液比1:8加入体积分数为70%的乙醇水溶液中浸提70小时,浸提期间要不断搅拌,然后提取液先用滤布真空抽滤,再用滤纸真空抽滤,将滤液减压浓缩得粗提液。
将ab-8型大孔树脂用无水乙醇浸泡20h,浸泡期间须不断搅拌。之后,将树脂颗粒和无水乙醇一同装柱(柱体积为1l),并用无水乙醇洗至流出液加水(1:5)不浑浊,再用纯水冲洗至无醇味。将柱子内的纯水放至与树脂相平时,将1l粗提液缓慢加入到层析柱中,到柱子下层的树脂全部被染上颜色后开始接收有明显颜色的流出液(浅绿色、棕黄色),进液相分析,以免样品过多,大孔树脂吸附不完全,出现过载现象。待样品被完全吸附后,依次用水、体积分数为20%的乙醇水溶液、体积分数为50%的乙醇水溶液和体积分数为80%的乙醇水溶液进行洗脱,洗脱至无色(一直不变),同时用干净玻璃瓶接出流分(每400ml接一个流分)。将接出的流分编号为a1、a2、a3……。将收集的流分过0.45μm滤膜过滤,然后进行高效液相色谱检测。液相色谱检测条件:色谱柱型号为venusilmpc18,4.6mm×250mm,5μm;检测波长为205nm,流动相a为甲醇,流动相b为0.1%甲酸水,进样量20μl。洗脱梯度为:初始,流动相a与流动相b的体积比为5:95;至30分钟变化为100%甲醇。根据检测结果进行合并,选择活性组分a27-29进一步分离和纯化,浓缩冻干得到粗提物。
将rp-c18填料(40μm,
将预分离物用体积分数为30%的甲醇水溶液溶解,使用lc3000型高效液相色谱仪、venusilmpc18色谱柱(10mm×250mm,5μm)、以流动相a为甲醇、流动相b为体积分数0.1%的甲酸水溶液作为流动相,洗脱梯度为:初始-30分钟,流动相a与流动相b的体积比为30:70;31分钟-40分钟,流动相a与流动相b的体积比为100:0。控制进样量80μl、流速3ml/min、检测波长205nm,进行液相半制备,收集含有目标化合物的流分段,浓缩冻干得到目标化合物。
实施例3
将朝鲜蓟晒干并用粉碎机进行粉碎,在室温条件下,将朝鲜蓟粉末按料液比1:9加入体积分数为70%的乙醇水溶液中浸提71小时,浸提期间要不断搅拌,然后提取液先用滤布真空抽滤,再用滤纸真空抽滤,将滤液减压浓缩得粗提液。
将ab-8型大孔树脂用无水乙醇浸泡22h,浸泡期间须不断搅拌。之后,将树脂颗粒和无水乙醇一同装柱(柱体积为1l),并用无水乙醇洗至流出液加水(1:5)不浑浊,再用纯水冲洗至无醇味。将柱子内的纯水放至与树脂相平时,将1l粗提液缓慢加入到层析柱中,到柱子下层的树脂全部被染上颜色后开始接收有明显颜色的流出液(浅绿色、棕黄色),进液相分析,以免样品过多,大孔树脂吸附不完全,出现过载现象。待样品被完全吸附后,依次用水、体积分数为20%的乙醇水溶液、体积分数为50%的乙醇水溶液和体积分数为80%的乙醇水溶液进行洗脱,洗脱至无色(一直不变),同时用干净玻璃瓶接出流分(每400ml接一个流分)。将接出的流分编号为a1、a2、a3……。将收集的流分过0.45μm滤膜过滤,然后进行高效液相色谱检测。液相色谱检测条件:色谱柱型号为venusilmpc18,4.6mm×250mm,5μm;检测波长为205nm,流动相a为甲醇,流动相b为0.1%甲酸水,进样量20μl。洗脱梯度为:初始,流动相a与流动相b的体积比为5:95;至30分钟变化为100%甲醇。根据检测结果进行合并,选择活性组分a27-29进一步分离和纯化,浓缩冻干得到粗提物。
将rp-c18填料(50μm,
将预分离物用体积分数为30%的甲醇水溶液溶解,使用lc3000型高效液相色谱仪、venusilmpc18色谱柱(10mm×250mm,5μm)、以流动相a为甲醇、流动相b为体积分数0.1%的甲酸水溶液作为流动相,洗脱梯度为:初始-30分钟,流动相a与流动相b的体积比为30:70;31分钟-40分钟,流动相a与流动相b的体积比为100:0。控制进样量80μl、流速3ml/min、检测波长205nm,进行液相半制备,收集含有目标化合物的流分段,浓缩冻干得到目标化合物。
对实施例1-3得到的化合物单体均进行lc-ms测定和核磁共振检测。
如图3所示,m/z339.0928[m+na]+,推测其,分子式为c15h21clo5。
对h谱(见图4)和c谱(见图5)的峰进行归属:
1h-nmr中,δ4.90处的峰为水峰;δ5.01处为一组单峰,通过积分面积得知包含有两个质子,因此推断为烯烃质子信号;δ1.20为一组甲基信号;高场部分的信号应该为多组-ch2-基团质子信号。13c-nmr中,δ48附近的峰为溶剂峰(氘代甲醇)。除去溶剂峰,谱图中共有15组碳信号,该结论与质谱结论相一致。
对h谱和c谱的峰进行归属如下:
1h-nmr:2.82(1h,f,j=6.0hz,h-1),2.03(1h,m,h-2a),1.69(1h,f,j=11.0hz,h-2b),3.63(1h,dt,j=10.0hz,5.0hz,h-3),1.77(1h,m,h-4),1.96(1h,f,j=9.0hz,h-5),4.15(1h,t,j=10.0hz,h-6),2.39(1h,t,j=10.0hz,h-7),3.97(1h,dt,j=10.0hz,4.0hz,h-8),2.77(1h,dd,j=12.0hz,4.0hz,h-9a),2.08(1h,t,j=11.0hz,h-9b),4.08(1h,d,j=11.0hz,h-13a),3.72(1h,d,j=11.0hz,h-13b),5.01(2h,s,h-14),1.20(3h,d,j=6.0hz,h-15)。
13c-nmr:41.61(c-1),37.66(c-2),77.14(c-3),46.41(c-4),51.29(c-5),81.03(c-6),60.18(c-7),69.64(c-8),46.59(c-9),143.81(c-10),77.53(c-11),176.66(c-12),42.49(c-13),113.56(c-14),17.41(c-15)。
本化合物的h-hcosy(见图6)谱图十分清晰,除δ5.01为一组单峰外,其它信号全部有耦合裂分情况,这给解析带来很大的帮助。hsqc(见图7)中,δ42.49分别与δ4.08(h,d,j=10.0hz)以及3.72(h,d,j=11.0hz)对应;δ46.59分别与δ2.77(h,dd,j=12.0hz/4.0hz)以及2.08(h,d,j=11.0hz)对应;δ37.66分别与δ2.03(h,m)以及1.69(h,f,j=11.0hz)对应,说明该三组信号为-ch2-基团。结合h-hcosy、hsqc,通过hmbc(见图8)可确定不同片段的连接位点,综合所有核磁谱图及质谱结果,化合物鉴定为cynarininb(愈创木烷倍半萜内酯的一种)。目标化合物结构式为:
本申请提供的从朝鲜蓟中提取愈创木烷型倍半萜的方法,从朝鲜蓟中分离提取出了目标化合物,方法简单实用,适宜于规模化应用,产品纯度高,对于朝鲜蓟的深度开发利用有着积极意义。
尽管已用具体实施例来说明和描述了本发明,然而应意识到,在不背离本发明的精神和范围的情况下可以作出许多其它的更改和修改。因此,这意味着在所附权利要求中包括属于本发明范围内的所有这些变化和修改。
- 还没有人留言评论。精彩留言会获得点赞!