一种3-羰基吲哚啉类荧光分子的合成方法及应用与流程
本发明属于有机合成领域,具体涉及一种3-羰基吲哚啉类荧光分子的合成方法及应用。
背景技术:
荧光分子是一类在紫外-可见-近红外区有特征荧光,并且其荧光性质(激发和发射波长、强度、寿命、偏振等)可随所处环境的性质,如极性、折射率、粘度等改变的一类荧光性分子。它们在生命科学研究领域,荧光染料在生物活体中成像、以及生物分子标记、示踪等方面已经得到了广泛的应用。在医学诊断领域,荧光分子也广泛使用于肿瘤成像等关键诊断方面,因此具有极高的实际应用价值。
3-羰基吲哚啉类化合物就是一类重要的荧光分子。由于这类分子内存在刚性共轭电子给体-供体片段,限制了分子内的旋转从而表现出一些荧光性质。这类荧光分子可以与一些荧光较弱的生物分子结合来可视化这些分子,因而在生物分子传感、运转或组织成像方面起到了极大的作用。
因此,针对3-羰基吲哚啉类荧光分子,从简单易得的原料出发,开发简便且廉价的合成方法具有重要的意义和极高的实际应用价值。在此之前,3-羰基吲哚啉类荧光分子的合成已有一些报道。例如,2003年刘亚华和williamw.mcwhorter课题组利用格式试剂将酮转化为醇再重排生成酮。但是其缺点是格式试剂具有一定的危险性且不易保存,一般都需要现制现用。(j.org.chem,vol.68,no.7,2003)
2013年yogeshgoriya和chepuriv.ramana课题组以邻溴苯甲酮与叠氮化钠为原料,通过snar-smalley环化反应得到与本发明类似产物。但是此方法所使用的叠氮化钠是剧毒品,可能与碱土金属、一价或多价的重金属盐,特别是铜、铅等等反应,生成爆炸性的重金属叠氮化物。危险性非常高,且不易后处理。(chem.commun.2013,49,6376)。
以上两种方法合成的此类分子均不具荧光特性,因此科学家又对具有类似结构且具有荧光性质的分子进行了探究。
化学家kaushik和apurbalalkoner在2017年,以二氢吲哚-2,3-二酮为起始原料,通过格式反应合成了类似分子。因此这个合成路线需要使用氢化钠、格氏试剂等较危险且易变质的试剂,而且合成过程对温度要求苛刻,比如第一步需要保持低温,而且第二步需要加热回流(chem.eur.j.2017,23,8610–8614)。
2018年mathieuachard课题组利用钌催化剂对取代吲哚进行烯丙基化,再氧化得到与本发明类似产物。但是此方法对反应条件特别苛刻,第一步中如果催化剂和溶剂的量加的不准确就会导致选择性和转化率特别低。(chem.eur.j.2018,24,7964–7969)
技术实现要素:
本发明旨在提供一种新的合成荧光分子的途径及应用,新的合成途径,转化率高、危险性低,易于操作。具体方案如下:
一种3-羰基吲哚啉类荧光分子的合成方法,式i所示的卤代苯甲醛类化合物与式ii所示的亚胺类化合物经两步催化后再进行氧化合成3-羰基吲哚啉类荧光分子:
其中,x为卤素;
ar为苯基、取代苯基或萘;r1为苯基、取代苯基、苄基、取代苄基、吡啶基以及烯丙基;r2为苯基或甲基;
其中第一步催化步骤所采用的催化剂为光敏催化剂,同时加入胺类化合物;第二步催化所采用的催化剂为金属催化剂。胺类化合物在此反应中充作电子供体。
优选的,所述的ar为取代苯基时,取代苯基的取代基为1-10个碳原子的烷基或1-10个碳原子的烷氧基;或
所述的r1为取代苯基时,取代苯基的取代基为卤素;r1为取代苄基时,取代基为1-10个碳原子的烯基基团;r1为官能化烷基时,为具有1-10个碳原子的烯基官能化的烷基。
优选的,式i所述的卤代苯甲醛为邻溴代苯甲醛;式ii所述的亚胺类化合物选自如下:
优选的,所述的合成方法的具体步骤为:
(1)采用式i所示的卤代苯甲醛类化合物与式ii所示的亚胺类化合物在光敏催化剂、胺类化合物、光源及有机溶剂的存在下反应得到中间产物;
(2)步骤(1)得到的中间产物在碱、有机溶剂的存在下经催化剂催化偶联后,再采用氧化剂进行氧化即得;即步骤(2)实际包含两个小步骤:1)催化偶联的步骤和2)氧化的步骤。
步骤(1)所述的光敏催化剂为有机光敏催化剂或金属光敏催化剂,金属光敏催化剂为钌催化剂或铱催化剂;所述的胺类化合物包括甲胺类化合物、乙胺类化合物、芳基胺类化合物;所述的光源为可见光;胺类化合物在此反应中起供电子的作用。
步骤(2)所述的碱为碱金属或碱土金属;催化剂为金属催化剂和有机磷配体配位的组合物,所述的金属催化剂为镍、铜、银、金、钯或铑催化剂;所述的氧化剂为氧气、三氟甲烷磺酸铜、n-溴代丁二酰胺或叔丁基过氧化氢或dmp;此处的dmp即dess-martin氧化剂。
步骤(1)的催化步骤和步骤(2)中的催化、氧化步骤采用的有机溶剂选自醇类、酯类、苯类、酰胺类、亚砜类、呋喃类、腈类化合物。
优选的,步骤(1)中所述的光敏催化剂选自如下结构:
或
步骤(1)所述胺类化合物为二甲胺、三甲胺、二乙胺、三乙胺、苯胺、己二胺、二异丙基甲胺、二异丙基乙胺、二环己基乙胺、n,n-二甲基苯胺、二芳基胺、三芳基胺、二环己基甲胺;或
所述的光源为太阳光、led光;或
步骤(2)所述的碱为碳酸钠、碳酸氢钠、碳酸钾、碳酸镁、碳酸铯。
进一步优选的,步骤(1)所述的光敏催化剂为:
所述的胺类化合物为二环己基甲胺、二异丙基乙胺、三乙胺;所述的光源为20w的蓝色led光;
步骤(2)所述的碱为碳酸铯;所述的金属催化剂为三(二亚苄基茚丙酮)二钯)、双(二亚芐基丙酮)钯、四(三苯基膦)钯、双三苯基磷二氯化钯;所述的氧化剂为dmp或氧气;或
所述的有机磷配体为三(邻甲基苯基)磷、甲基三苯基溴化膦或三苯基膦;或
步骤(1)采用的有机溶剂为乙腈;步骤(2)偶联步骤采用的有机溶剂为甲苯;步骤(2)的氧化步骤采用的有机溶剂为二氯甲烷。
进一步优选的,步骤(1)是在无水无氧环境下进行的。
进一步优选的,所述的亚胺类化合物与卤代苯甲醛类化合物的摩尔比为1:1~3;光敏催化剂与亚胺类化合物的摩尔比为0.5%~1%;胺类化合物与亚胺类化合物的摩尔比为1~3;或所述的步骤(1)的反应温度为0℃~50℃;步骤(2)中偶联反应为温度为80~110℃;氧化反应步骤的温度为0℃~30℃。
进一步优选的,所述的亚胺类化合物与卤代苯甲醛类化合物的摩尔比为1:1.2;光敏催化剂与亚胺类化合物的摩尔比为0.5%;胺类化合物与亚胺类化合物的摩尔比为2;或
步骤(1)的反应温度为25℃;步骤(2)中偶联反应为温度为110℃;氧化反应步骤的温度为25℃。
根据上述任一项所述的合成方法合成的产物在荧光标记中应用。
采用本发明方法,具有如下至少一种有益效果:
本发明由醛和亚胺在光催化体系中反应得到的产物经过金属催化的偶联反应制备的氨基醇产物易于转化为生物荧光探针或生物活性物质,开发了一种新的合成荧光分子的途径。本发明方法原料便宜易得;本发明的方法反应条件极度温和,无需采用极端温度等条件;反应易于操作;反应所得产物的产率很高,有意料不到的好效果;反应步骤少,两步反应仅需最后一步纯化即可得到目标荧光分子;目标荧光分子的结构很容易进行微调,从而产生不同的荧光光谱,并且可以形成不同的官能团从而使其应用更加广泛。
附图说明
图1为实施例1中产物的1hnmr核磁分析谱图。
图2为实施例1中产物的13cnmr核磁分析谱图。
图3为实施例1中产物的荧光分析谱图。
图4为实施例2中产物的1hnmr核磁分析谱图。
图5为实施例2中产物的13cnmr核磁分析谱图。
图6为实施例2中产物的荧光分析谱图。
图7为实施例3中产物的1hnmr核磁分析谱图。
图8为实施例3中产物的13cnmr核磁分析谱图。
图9为实施例3中产物的荧光分析谱图。
图10为实施例4中产物的1hnmr核磁分析谱图。
图11为实施例4中产物的13cnmr核磁分析谱图。
图12为实施例4中产物的荧光分析谱图。
图13为实施例5中产物的1hnmr核磁分析谱图。
图14为实施例5中产物的13cnmr核磁分析谱图。
图15为实施例5中产物的荧光分析谱图。
图16为实施例6中产物的1hnmr核磁分析谱图。
图17为实施例6中产物的13cnmr核磁分析谱图。
图18为实施例6中产物的荧光分析谱图。
图19为实施例7中产物的1hnmr核磁分析谱图。
图20为实施例7中产物的13cnmr核磁分析谱图。
图21为实施例8中产物的1hnmr核磁分析谱图。
图22为实施例8中产物的13cnmr核磁分析谱图。
图23为实施例12的荧光成像分析结果图
具体实施方式
实施例中所使用的所有化学试剂除了亚胺是现场合成(合成方法均为现有技术),其他原料均购自市场。以下实施例中所使用的仪器包括400兆核磁波谱仪(jnm-ecz400s)以及荧光光谱仪爱登堡(ei-fls980)。
通过dess-martin氧化以氨基醇为原料的荧光分子的合成
将亚胺(0.2mmol),光催化剂(1-3毫克,0.5-1.5mmol%),胺(42.8-128.4微升,0.4mmol,1-3eq),邻溴苯甲醛(0.2-0.6mmol,1-3eq)在无水无氧氛围下溶于有机溶剂中,室温下可见光照射搅拌反应12-24小时。用旋转蒸发仪除去挥发性溶剂,将得到的粗产物(0.1mmol),金属催化剂(5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,1.3eq)溶解在有机溶剂中,加入到干燥的带有磁子的史莱克管中。然后在搅拌的情况下将该反应体系置于预热的油浴(80-110℃)下反应约10-24小时。反应结束后,将反应混合物冷却至室温,然后打开至空气,并通过硅藻土过滤,用乙酸乙酯(3x2毫升)冲洗硅藻土,收集滤液,将滤液使用旋转蒸发仪除去所有挥发性溶剂,得到的粗产品直接用于下一步氧化步骤。将粗产品(0.1mmol)溶于二氯甲烷中,冷却后将氧化剂缓慢加入至此反应液中,将所得溶液在室温下搅拌20-50分钟,反应结束后及时使用旋转蒸发仪除去溶剂,并通过柱层析法用石油醚和乙酸乙酯作为洗脱剂分离得到目标荧光产物。
实施例1
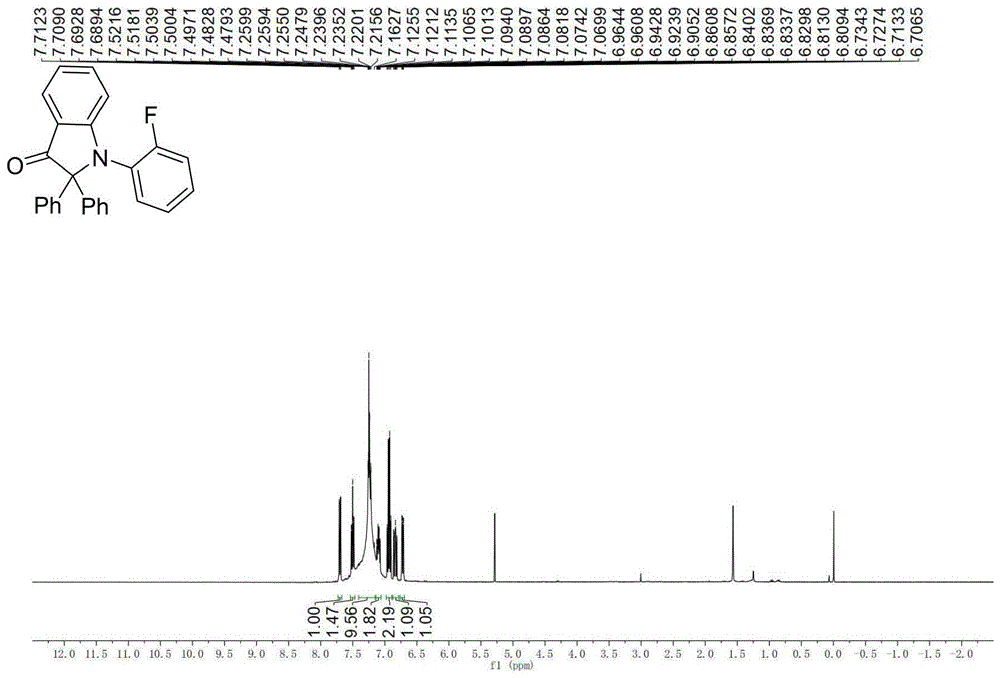
按照典型实验过程,将n-(2-氟苯基)-1,1-二苯基甲亚胺(0.2mmol),光催化剂ir-9(1毫克,0.5mmol%),二环己基甲胺(85.6微升,0.4mmol,2eq),邻溴苯甲醛(0.24mmol,1.2eq)在无水无氧的氛围下溶于乙腈(2毫升)中,25℃下用20w的蓝色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得粗产物(0.1mmol),三(二亚苄基茚丙酮)二钯)(4.6毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.13mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应约12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(21毫克,产率:56%)。产物结构如上所示,产物的1hnmr核磁分析谱图、13cnmr核磁分析谱图、荧光分析谱图分别如图1-3所示,产物的测试数据为:(ir:1708,1601,1498,1475,1464,1445,1356,1325,1304,1266,1299,1151,1108,1017,940,930,809,751,704,697,636,612,524,491cm-1;hrms:计算量为:380.1451,实测量为:380.1450[m+h]+)。1hnmr(400mhz,chloroform-d):δ7.70(dd,j=7.8,1.3hz,1h),7.50(ddd,j=8.4,7.1,1.4hz,2h),7.35–7.15(m,9h),7.10(m,2h),6.93(q,j=7.4hz,2h),6.84(ddd,j=11.0,8.2,1.5hz,1h),6.72(dd,j=8.4,2.7hz,1h).13cnmr(101mhz,chloroform-d)δ200.81,160.55,160.34,158.03,137.70,129.95,129.70–128.80(m,j=13.8hz),128.58(d,j=8.06hz),128.21,127.19(d,j=11.18hz),125.52,124.40,124.37,119.99,119.36,116.73,116.52,111.91,111.89,83.06.荧光数据(甲醇溶液):最大激发波长:407nm,最大发射波长:503nm)
实施例2
按照典型实验过程,将1,1-二苯基-n-吡啶基亚胺(0.2mmol),光催化剂ir-9(1毫克,0.5mmol%),二环己基甲胺(85.6微升,0.4mmol,2eq),邻溴苯甲醛(0.24mmol,1.2eq)在氮气氛围下溶于乙腈(2毫升)中25℃下用20w的蓝色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),三(二亚苄基茚丙酮)二钯)(4.6毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.13mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应约12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(14毫克,产率:40%)。产物的1hnmr核磁分析谱图、13cnmr核磁分析谱图、荧光分析谱图分别如图4-6所示,产物的测试数据为:(ir:1705,1612,1578,1482,1464,1417,1351,1326,1191,1016,924,802,710,702cm-1;hrms:计算量:363.1497,实测量:363.1497,[m+h]+)。1hnmr(400mhz,chloroform-d):δ8.41(d,j=2.6hz,1h),8.23(dd,j=4.7,1.5hz,1h),7.71(dd,j=7.8,1.3hz,1h),7.53(ddd,j=8.5,7.1,1.4hz,1h),7.40–7.30(m,5h),7.29–7.22(m,6h),7.10(d,j=8.4hz,1h),7.03(dd,j=8.3,4.7hz,1h),6.97(t,j=7.4hz,1h).13cnmr(101mhz,chloroform-d):δ200.82,158.93,147.07,146.49,137.80,137.71,137.08,132.45,129.09,128.59,128.51,126.05,123.50,120.75,120.60,111.79,82.86.荧光数据(甲醇溶液):最大激发波长:405nm,最大发射波长:495nm)
实施例3
按照典型实验过程,将n-烯丙基-1,1-双苯基亚胺(0.2mmol),光催化剂ir-9(1毫克,0.5mmol%),二环己基甲胺(85.6微升,0.4mmol,2eq),邻溴苯甲醛(0.24mmol,1.2eq)在无水无氧的氛围下溶于乙腈(2毫升)中,25℃下用20w的蓝色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),三(二亚苄基茚丙酮)二钯)(4.6毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应约12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(15毫克,产率:45%)。产物的1hnmr核磁分析谱图、13cnmr核磁分析谱图、荧光分析谱图分别如图7-9所示,产物的测试数据为:(ir:3061,3024,2925,2856,1697,1615,1576,1486,1464,1444,1414,1375,1322,1301,1275,1191,1165,1088,1015,995,864,679,605,545,520cm-1,hrms:计算量:326.1545,实测量:326.1543,[m+h]+)。1hnmr(400mhz,chloroform-d):δ7.65–7.56(m,1h),7.48(ddd,j=8.4,7.1,1.4hz,1h),7.37–7.16(m,10h),6.82(d,j=8.3hz,1h),6.79–6.72(m,1h),5.06(ddt,j=17.1,9.8,5.4hz,1h),4.98–4.83(m,2h),4.03(dt,j=5.4,1.5hz,2h).13cnmr(101mhz,chloroform-d):δ200.96,159.98,138.87,137.88,132.95,128.71,128.67,128.26,125.73,118.98,117.74,117.34,109.31,80.22,46.78.荧光数据(甲醇溶液):最大激发波长:398nm,最大发射波长:530nm)
实施例4
按照典型实验过程,将1-萘基-1-甲基-n-苯基亚胺(0.2mmol),光催化剂ir-9(1毫克,0.5mmol%),二环己基甲胺(85.6微升,0.4mmol,2eq),邻溴苯甲醛(0.24mmol,1.2eq)在无水无氧的氛围下溶于乙腈(2毫升)中,25℃下用20w的蓝色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),三(二亚苄基茚丙酮)二钯)(4.6毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应约12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(14毫克,产率:40%)。产物的1hnmr核磁分析谱图、13cnmr核磁分析谱图、荧光分析谱图分别如图10-12所示,产物的测试数据为:(ir:3065,2925,1695,1606,1573,1497,1480,1464,1360,1325,1185,1145,1100,1065,1030,994,960,928,906,863,789,773,636,544,477cm-1,hrms:计算量:350.1545,实测量:350.1544,[m+h]+)。1hnmr(400mhz,chloroform-d):δ7.87(t,j=8.0hz,3h),7.59–7.49(m,2h),7.46–7.32(m,3h),7.28–7.21(m,1h),7.16–7.10(m,3h),7.08–6.96(m,2h),6.71(dd,j=6.8,2.9hz,2h),1.72(s,3h).13cnmr(101mhz,chloroform-d):δ202.89,157.95,139.03,137.26,134.42,134.28,131.48,130.05,129.30,129.18,127.92,126.96,126.91,126.74,125.83,125.61,125.11,124.82,120.08,119.51,112.19,74.60,23.00.荧光数据(甲醇溶液):最大激发波长:422nm,最大发射波长:495nm)
实施例5
按照典型实验过程,将n-苄基-1-苯基-1-(对甲苯基)亚胺(0.2mmol),光催化剂ir-9(1毫克,0.5mmol%),二环己基甲胺(85.6微升,0.4mmol,2eq),邻溴苯甲醛(0.24mmol,1.2eq)在无水无氧的氛围下溶于乙腈(2毫升)中,25℃下用20w的蓝色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),三(二亚苄基茚丙酮)二钯)(4.6毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应约12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(30毫克,产率:75%)。产物的1hnmr核磁分析谱图、13cnmr核磁分析谱图、荧光分析谱图分别如图13-15所示,产物的测试数据为:(ir:3059,3027,2921,1705,1611,1507,1485,1452,1382,1353,1163,1029,921,868,749,634,545,518,489,454cm-1,hrms:计算量:390.1858,实测量:390.1858,[m+h]+)。1hnmr(400mhz,chloroform-d):δ7.65(dd,j=7.7,1.3hz,1h),7.39(ddd,j=8.5,7.1,1.4hz,1h),7.30(m,5h),7.18(d,j=8.3hz,2h),7.15(m,5h),6.75(m,3h),6.60(d,j=8.3hz,1h),4.70(s,2h),2.32(s,3h).13cnmr(101mhz,chloroform-d):δ200.81,160.11,138.83,138.07,137.95,137.29,135.52,129.51,128.97,128.85,128.76,128.37,128.20,126.92,126.64,125.80,119.02,117.95,109.27,80.89,48.12,21.19.荧光数据(甲醇溶液):最大激发波长:420nm,最大发射波长:498nm)
实施例6
按照典型实验过程,将n-苄基-1-苯基-1-(对甲氧苯基)亚胺(0.2mmol),光催化剂ir-9(1毫克,0.5mmol%),二环己基甲胺(85.6微升,0.4mmol,2eq),邻溴苯甲醛(0.24mmol,1.2eq)在无水无氧的氛围下溶于乙腈(2毫升)中,25℃下用20w的蓝色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),三(二亚苄基茚丙酮)二钯)(4.6毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应约12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(25毫克,产率:63%)。产物的1hnmr核磁分析谱图、13cnmr核磁分析谱图、荧光分析谱图分别如图16-18所示,产物的测试数据为:(ir:3060,2927,1704,1610,1579,1467,1453,1354,1321,1252,1178,1031,967,868,793,573,518cm-1,hrms:计算量:405.1729,实测量:405.1730,[m+h]+)。1hnmr(400mhz,chloroform-d):δ7.64(ddd,j=7.7,1.4,0.6hz,1h),7.38(ddd,j=8.5,7.1,1.4hz,1h),7.32–7.27(m,5h),7.20(d,j=8.9hz,2h),7.08(dd,j=5.0,2.0hz,3h),6.85–6.79(m,2h),6.79–6.72(m,3h),6.59(dd,j=8.3,0.8hz,1h),4.69(d,j=3.2hz,2h),3.76(s,3h).13cnmr(101mhz,chloroform-d):δ200.92,160.07,159.55,138.79,137.96,137.27,130.54,130.23,128.88,128.77,128.38,128.20,126.94,126.62,125.80,118.93,117.93,114.20,109.23,80.60,55.41,48.05.荧光数据(甲醇溶液):最大激发波长:419nm,最大发射波长:500nm)
实施例7
按照典型实验过程,将n-苄基-1,1-二苯基亚胺(0.2mmol),光催化剂ir-9(1毫克,0.5mmol%),二环己基甲胺(85.6微升,0.4mmol,2eq),邻溴苯甲醛(0.24mmol,1.2eq)在无水无氧的氛围下溶于乙腈(2毫升)中,25℃下用20w的蓝色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),三(二亚苄基茚丙酮)二钯)(4.6毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应约12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(22.5毫克,产率:60%)。产物的1hnmr核磁分析谱图、13cnmr核磁分析谱图分别如图19-20所示,产物的测试数据为:(ir:3380,3056,3025,3004,1705,1611,1492,1477,1464,1342,1151,1057,1019,1019,932,753,694cm-1;hrms计算量:376.1701,实测量:376.1689[m+h]+.1hnmr(400mhz,chloroform-d)δ7.65(d,j=7.4hz,1h),7.44–7.39(m,1h),7.34–7.28(m,10h),7.09–7.01(m,3h),6.77(t,j=7.4hz,1h),6.72(dd,j=7.2,1.9hz,2h),6.60(d,j=8.3hz,1h),4.71(s,2h)ppm;13cnmr(101mhz,chloroform-d)δ200.67,160.15,138.63,138.06,137.17,128.96,128.82,128.64,128.52,128.38,128.28,127.48,127.17,126.97,126.57,125.84,118.96,118.05,109.28,81.05,48.12)
实施例8
按照典型实验过程,将1,1-二苯基-n-(4-乙烯基苄基)亚胺(0.2mmol),光催化剂ir-9(1毫克,0.5mmol%),二环己基甲胺(85.6微升,0.4mmol,2eq),邻溴苯甲醛(0.24mmol,1.2eq)在无水无氧的氛围下溶于乙腈(2毫升)中,25℃下用20w的蓝色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),三(二亚苄基茚丙酮)二钯)(4.6毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应约12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(20.9毫克,产率:52%)。产物的1hnmr核磁分析谱图、13cnmr核磁分析谱图分别如图21-22所示,产物的测试数据为:(ir:3485,3059,2919,2849,1707,1610,1486,1320,998,867,750,701,603,487cm-1;hrms计算量:402.1858,实测量:402.1859[m+h]+.1hnmr(400mhz,chloroform-d)δ7.65(d,j=7.3hz,1h),7.42–7.36(m,1h),7.32–7.27(m,10h),7.11(d,j=8.2hz,2h),6.77(t,j=7.4hz,1h),6.67(d,j=8.2hz,2h),6.62–6.53(m,2h),5.63(d,j=17.7hz,1h),5.16(d,j=11.0hz,1h),4.69(s,2h)ppm;13cnmr(101mhz,chloroform-d)δ200.63,160.06,138.66,138.05,136.80,136.42,136.36,128.94,128.85,128.31,126.78,126.26,125.84,119.00,118.10,113.74,109.32,81.05,47.98)
实施例9
按照典型实验过程,将n-(2-氟苯基)-1,1-二苯基甲亚胺(0.2mmol),光催化剂ir-5(1毫克,0.5mmol%),二异丙基乙胺(66微升,0.4mmol,2eq),邻溴苯甲醛(0.2mmol,1eq)在无水无氧的氛围下溶于四氢呋喃中,40℃下用太阳光照射搅拌反应约24小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),双三苯基磷二氯化钯(7.0毫克,5mol%),三(邻甲基苯基)磷(3毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴80℃下反应约24小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),30℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(7毫克,产率:17%)。(在265nm紫外灯下可观察到发光的目标产物荧光分子)
实施例10
按照典型实验过程,将1,1-二苯基-n-吡啶基亚胺(0.2mmol),光催化剂2(1.5毫克,1mmol%),三乙胺(83.4微升,0.6mmol,3eq),邻溴苯甲醛(0.3mmol,1.5eq)在氮气氛围下溶于n,n-二甲基甲酰胺中,20℃下用20w的绿色led光照射搅拌反应约20小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),双(二亚芐基丙酮)钯(5.7毫克,5mol%),甲基三苯基溴化膦(7.1毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴100℃下反应约20小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢通入氧气。30℃下反应约50分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(7毫克,产率:20%)。(在265nm紫外灯下可观察到发光的目标产物荧光分子)
实施例11
按照典型实验过程,将n-烯丙基-1,1-双苯基亚胺(0.2mmol),光催化剂ru-3(1毫克,0.5mmol%),二环己基甲胺(42.8微升,0.2mmol,1eq),邻溴苯甲醛(0.6mmol,3eq)在无水无氧的氛围下溶于二甲基亚砜中,50℃下用20w的红色led光照射搅拌反应约24小时。用旋转蒸发仪除去挥发性溶剂,将所得的粗产物(0.1mmol),四(三苯基膦)钯(11.5毫克,5mol%),三苯基膦(5.2毫克,10mol%),碳酸铯(42毫克,0.26mol)溶于甲苯。然后在搅拌的情况下将该反应体系置于预热的油浴110℃下反应12小时。反应结束后,通过硅藻土过滤并旋干,将得到的粗产物(0.1mmol)用二氯甲烷溶解并冷却,缓慢加入dess-martin氧化剂(42毫克,0.1mmol),25℃下反应约30分钟,反应结束以后旋干溶剂,通过柱层析法分离得到目标荧光分子(6毫克,产率:18%)。(在265nm紫外灯下可观察到发光的目标产物荧光分子)。
实施例12实施例应用
在一个干净平整的96孔微板中加入牛血清蛋白(bsa)(100微升,15μm,1eq),接着加入实施例8的产物(6b)的二甲基亚砜(dmso)溶液(1.5微升,20mm,20eq)。将该反应体系置于365nm的紫外光下照射约0.5小时。在此,我们做了几组对照实验:1)不加6b也不用紫外光照射;2)不加6b;3)加入6b但不用紫外光照射;4)加入6b同时用紫外光照射。随后对这四个蛋白进行凝胶电泳分析,首先考马斯蓝染色结果证明每一个条带都含有bsa蛋白。而荧光成像分析结果如图23所示,显示只有第四组条带的蛋白显示出荧光信号,证明荧光分子6b在紫外光下可以特异性的对bsa进行标记。
- 还没有人留言评论。精彩留言会获得点赞!