核酸扩增装置、核酸扩增方法以及核酸扩增用芯片与流程
本申请是基于申请号201580036603.x、发明名称为“核酸扩增装置、核酸扩增方法以及核酸扩增用芯片”的发明专利申请的分案申请。上述发明专利申请为申请号pct/jp2015/069549的pct国际申请的中国国家阶段申请,申请日为2015年7月7日。
本发明涉及一种核酸扩增装置、核酸扩增方法以及核酸扩增用芯片。
背景技术:
核酸的检测是药物研发、法医学、临床试验、农产品和病原微生物类型鉴定等诸多领域的核心。通过分子系统发育分析而能够检测诸如癌症、微生物感染和基因标记等各种疾病是疾病和疾病发展风险的诊断、标记搜索、食品和环境安全评价、犯罪证据以及许多其它技术方面的通用技术。
用于检测少量核酸(基因)的最强大的基础技术之一为一种用于对通过核酸序列的部分或全部进行呈指数地复制而获得的产物进行分析的方法。
pcr是一种对dna的特定区域进行选择性扩增的有效技术。利用pcr,可以从单一模板dna分子快速地在模板dna中复制产生数百万拷贝的目的dna序列。在pcr中,重复进行两相或三相温度循环(称为“热循环”)从而依次进行以下各个反应:变成单链的dna变性;形成变性的单链dna的引物退火;以及使用耐热dna聚合酶的引物延伸。重复该循环直至得到用于分析的足够拷贝数量。原理上,pcr的每次循环可使拷贝数量增加一倍。实际上,随着热循环的继续,所需反应物的浓度降低,使得扩增的dna产物的构建最终停止。关于pcr大致细节,参见《pcr的临床应用》(dennislo主编,于1998年由位于totowa,newjersey的humana出版社出版)和《pcr协议-方法和应用指南》(m.a.innis等主编,于1990年由位于sandiego,california的学术出版社有限公司出版)。
虽然pcr是一种对期望dna进行选择性扩增的有效技术,而在完成pcr后有必要通过凝胶电泳等进行确认,用以对扩增的dna进行确定。因此,作为一种改进的pcr方法,开发了一种根据期望dna的扩增量的实时pcr进化或荧光淬灭技术,从而使样品中期望dna的存在与否变得容易确认。在传统pcr方法中,当在pcr之前样品中模板dna的量超出一定量时,pcr之后扩增的dna的量通常进入平稳状态,而pcr之前的模板dna的量无法进行定量。而在实时pcr方法中,在pcr进入平稳状态之前,可以对pcr过程中扩增的dna的量进行实时检测;因此,可根据dna的扩增状态来对pcr之前的模板dna的量进行定量。因此,实时pcr方法也被称之为定量pcr方法。
利用实时pcr的目的dna量的定量具有特殊的临床效用,例如,用于监测病毒载量的变化,用以确认对诸如艾滋病病毒(hiv)等的病毒感染的治疗效果。利用实时pcr的dna定量对机会性感染的诊断也是有效的,比如疱疹病毒(hhv),许多患者自从婴儿期就已经亚临床感染并由于较弱的体质等原因而发展。
pcr和实时pcr是通过热循环对基因进行呈指数地扩增的有效方法。用于pcr的通用热循环装置由于用作加热器的铝块的热容量较大,因而在温度控制方面较为缓慢,通常需要1到2小时、甚至在某些情况下需要更长时间才能进行pcr的30-40次循环。因此,即使使用现有最先进的基因测试装置,分析过程往往需要1小时以上。自从该技术开发以来,增加pcr的速度一直是一项巨大的挑战。业已开发了用于提高速度的各种方法。可将提高热循环样品的速度的方法分为以下三种类型。
在第一种方法中,样品溶液被导入装置中,在一定时间内进行温度循环并且使溶液保持在同一部位(非专利文献(npl)1和专利文献(ptl)1)。该方法意于通过减少样品的量而降低热容量,从而增加热循环的速度。然而,容纳腔或加热器本身的热容量的减少有一定限度。因此,每次循环至少需要约30秒才能实现足够的扩增反应。即使使用最快的装置,完成pcr反应也需要15分钟以上。
第二种方法被称之为连续流动式pcr。在该方法中,样品溶液通过微通道流经彼此隔开的多个温度区,同时不停地进行溶液的连续进样。在这种连续流动式pcr方法中,已知的系统是通过使样品溶液流经三个控制为恒温的加热器上方的蛇形微通道而快速控制样品温度的(npl2)。由于这种连续流动式pcr方法无需改变外部装置(如容器和加热器等)的温度,因此理论上可期望达到最快的温度控制。在极快的情况下,约7分钟之内完成dna扩增。然而,采用连续流动式pcr进行定量实时pcr,有必要使得能够对各个蛇形微通道的全区或在同一温度区的每个蛇形微通道的30-50个区域进行荧光观察。具体地,需要可均匀地照射较大区域的激发光源和用于荧光观察的高灵敏度摄像机或行扫描器,因此必然导致系统结构体积较大且价格昂贵。
在第三种方法中,如第二种方法,彼此间隔的多个温度区通过微通道而连接,并且使样品溶液往复地(reciprocally)流经相同微通道,且使得样品溶液在每个温度区停留一定时间段从而被加热(专利文献(ptl)2)。这种方法的优点在于进行热循环过程中样品与各温度区的接触时间可以自由设置。然而,为了导入样品并将其往复或旋转地泵至这些温度区,需要使用许多集成阀和泵以及用于观察样品溶液位置的检测器,用于防止样品溶液由于高温侧加热的样品中形成的小气泡的膨胀或由于在气液界面上形成的气压差(当样品溶液流经具有从用于变性反应95℃以上至用于退火反应约60℃的温度梯度的微通道时)而非意愿地流出微通道中的期望温度区位置,而且装置小型化困难(npl3、npl4和ptl3)。
使用pcr/实时pcr装置的基因检测市场在有利增长。尤其是诸如病毒性肝炎、性传播疾病和流感等传染性疾病的基因检测,在日本也迅速展开。基因检测对于癌症治疗的作用已变得明显。例如,egfr基因突变可用作施用癌剂iressa的一个经验法则。因此,对于肺癌、胰腺癌等中的egfr基因、k-ras基因,ews-flil基因、tls-chop基因,svt-ssx基因以及c-kit基因的基因检测最近已经进入保险的覆盖范围。
在pcr中,引物被连接至模板dna,并且位于引物序列之间的目的dna专门由dna聚合酶来检测。虽然pcr可用于检测dna,但pcr不能直接检测rna。因此,为了检测诸如流感病毒、诺如病毒(noroviruses)等rna病毒,在以rna为模板逆转录合成互补的cdna后再进行pcr;也就是进行所谓的rt-pcr。因此,实质上必须进行一个两阶段的步骤。此外,pcr和rt-pcr需要快速升温和快速降温,因而需要特殊的孵化器。因此,问题在于:使pcr和rt-pcr的应用自动化并不容易。
近年来,通过在一个pcr系统中使用多对引物而同时扩增多个基因区域的多重pcr引起人们的关注。从多重pcr发展而来的实时多重pcr,旨在受其他目的基因的影响(串扰)较小并且不损害灵敏度的条件下单独检测和定量多个不同的目的基因。然而,据报道,由于可标记的荧光物质波长重叠和种类问题使得两个以上的定量多重反应往往很难进行。
目前,基因检测是在实验室或分析中心进行的。然而,如果有可现场快速进行基因测试的高速实时pcr装置,则可以现场确定治疗方案和对策。因此,这样的装置被认为是可以取代目前的基因测试装置的一个划时代的技术。特别是,作为防止口蹄疫、高致病性流感之类的流行病的检疫措施,用于对其进行现场快速准确的判断并防止继发性感染的传播是非常重要的。因此,需要大量这样的高速实时pcr装置。
尤其是,为了实现在临床情景中或在发生传染病的现场立即进行基因测试的服务需求,需要一种低成本操作的、高速和高度便携式的实时pcr装置。
引用列表
专利文献
ptl1:公开号为2479452的加拿大专利申请
ptl2:jp2003-200041a
ptl3:wo2006/124458
非专利文献
npl1:neuzil等(实验室芯片,10:2632-2634(2010))
npl2:kopp等(科学,280:1046-1048(1998))
npl3:chiou等(分析化学,73:2018-2021(2001))
npl4:brunklaus等(电泳疗法,33:3222-3228(2012))。
技术实现要素:
技术问题
本发明的目的是提供一种能够现场使用并能进行高速实时pcr的小型核酸扩增装置、用于该装置的板以及核酸扩增方法。
解决问题的方案
为了实现了增加反应效率以及提供一种较小的扩增装置的目的,本发明提供了一种往复流动型(reciprocal-flow-type)高速实时核酸扩增装置,包括:设置在同一平面上的两个温度区;与各温度区接触的微通道,当鼓风机、风扇等停止时,所述微通道的两端设置为向大气压开放;以及液体输送机构,籍此活塞式样品溶液通过所述微通道在各个所述温度区的精确位置之间进行往复,从而进行热循环并同时确认pcr溶液的通过以及测量每次热循环的荧光强度。在另一个示例性实施例中,核酸扩增方法包括通过逆转录将rna转换为cdna以及将所述cdna进行pcr处理。
具体地,本发明提供了以下核酸扩增装置、核酸扩增方法、以及芯片。
(1)一种往复流动型核酸扩增装置,包括:
加热器,所述加热器能够形成变性温度区和延伸/退火温度区;
荧光检测器,所述荧光检测器能够检测样品溶液在两个温度区之间的移动;
一对液体输送机构,所述一对液体输送机构允许所述样品溶液在两个所述温度区之间移动,并且当液体输送停止时,所述液体输送机构设置为对大气压开放;
基板,核酸扩增用芯片可以放置在所述基板上;以及
控制机构,所述控制机构通过接收来自所述荧光检测器的与所述样品溶液的移动有关的电信号来控制各液体输送机构的驱动,
所述装置能够通过测量每次热循环的荧光强度来进行实时pcr。
(2)一种核酸扩增用芯片,包括至少一个微通道,所述微通道包括:
弯曲微通道,所述弯曲微通道分别用于根据(1)所述的核酸扩增装置的所述变性温度区和所述延伸/退火温度区;
直线中间微通道,所述直线中间微通道连接所述弯曲微通道;以及
连接部,所述连接部位于所述微通道的两端,
所述连接部可连接至根据(1)所述的核酸扩增装置的所述液体输送机构。
(3)根据(1)所述的核酸扩增装置,其中,所述液体输送机构为微型鼓风机或风扇。
(4)一种核酸扩增方法,包括以下步骤:
步骤1:将根据(2)所述的核酸扩增用芯片放置在根据(1)所述的基板上,使得所述变性温度区和所述延伸/退火温度区分别包括弯曲微通道;
步骤2:将样品溶液导入所述微通道中;
步骤3:将一对液体输送机构连接至位于所述微通道两端的液体输送机构连接部;以及
步骤4:利用所述液体输送机构将所述样品溶液在所述微通道的所述弯曲微通道之间进行往复从而进行热循环,并同时使用在所述中间微通道中的至少一个荧光检测器来测量每次热循环的所述样品溶液的荧光强度并确认所述样品溶液的通过,用以进行实时pcr。
(5)根据(4)所述的核酸扩增方法,其中,所述荧光强度的测量是通过同时测量两种以上荧光波长来同时测量在一个微通道中的多个基因的实时pcr而进行的。
(6)根据(4)或(5)所述的核酸扩增方法,其中,所述荧光强度的测量是通过使用从由每次热循环的荧光强度的矩阵(扩增曲线的二维数组)导出的循环次数ct求得的校准曲线而进行的。
(7)根据(4)至(6)任一项所述的核酸扩增方法,其中,所述核酸扩增方法选自由聚合酶链反应(pcr)、逆转录pcr(rt-pcr)、一步法pcr、多重pcr、多重rt-pcr、实时pcr以及实时rt-pcr组成的组。
(8)根据(4)到(7)任一项所述的核酸扩增方法,其中,所述核酸扩增方法包括执行中断分析,所述中断分析可通过在平坦的基板上形成两个以上微通道而进行,使得能够对通过各微通道的液体输送操作进行独立地控制。
(9)根据(4)至(8)任一项所述的核酸扩增方法,其中,所述方法包括:
将微量吸管的过滤吸头的端部连接至一个所述连接部,从而将样品溶液导入至所述微通道中;
在所述吸头与所述连接部相连接的状态下移除所述微量吸管,然后将所述吸头连接至一个所述液体输送机构。
(10)根据(4)至(9)任一项所述的核酸扩增方法,其中,导入至所述微通道中的所述样品溶液的体积在5μl-50μl的范围内。
(11)根据(2)所述的核酸扩增用芯片,所述芯片用于根据(4)至(10)任一项所述的核酸扩增方法中。
发明的有益效果
根据本发明,由于每个微通道设置有两个液体输送机构(优选风扇)和一个荧光检测器,因此实现了低成本、紧凑型的便携式装置。
此外,由于样品溶液在两个温度区之间往复,因此能够通过同时快速地确认pcr溶液的通过以及测量每次热循环的荧光强度,从而实现更快的实时pcr。
在使用对样品溶液进行往复的热循环的常规方法中,压力源(如注射器泵)连接至微通道,通过重复地加压和减压而使活塞状(plug-like)样品溶液进行往复。在此过程中,微通道在与压力源连接一侧的内部必须是一个封闭系统,以便不泄漏微通道中活塞状样品溶液的压力。当施加到气液界面的活塞状样品溶液的力(通过压力源施加或减少压力而产生)超过了活塞状样品溶液和微通道内壁之间的静摩擦力时,液体输送开始。另一方面,当停止加压或减压而使活塞状样品溶液停止时,作用在气液界面上的样品溶液的压力保持在压力源侧的封闭微通道之内,并且溶液保持移动一段时间直至能量完全被动摩擦消耗然后停止。尤其是,当样品溶液被加热到约95℃或更高进行变性反应(如在pcr中),内部压力的变化对样品溶液的影响很大(如粘度变化和小气泡的形成),压力源(比如泵)停止之后的移动量变化范围较大。为了使样品溶液停止在精确位置,有必要采取一些措施,如使用专用阀释放微通道的内部压力以及对压力源进行复杂操作,以及使用专用传感器来确认溶液的位置等。
相比之下,尽管本发明通过使用鼓风机、风扇等进行鼓风而在微通道中施加或减少压力以使活塞状样品溶液往复,当溶液达到各温度区上的精确位置或刚好达到各温度区上的精确位置之前,在鼓风机、风扇等的鼓风停止则液体输送也立即停止。这是因为当鼓风停止,微通道的内部压力瞬间对大气压开放、作用于活塞状溶液的压力消失,从而液体输送立即停止。因此,即使没有释放压力以控制样品溶液位置用的多个阀,也可以通过确认样品溶液仅在位于各温度区之间的一个位点处的通过即可实现精确的位置控制。此外,由于也可以同时在该位点(在此确认每次循环的往复液体的位置)使用荧光法,因此能够实现结构最简单的实时pcr热循环仪,其中只需使用直线微通道中的一个位点作为检测点。
在本发明中,使用聚合酶链反应(通常称之为pcr)。pcr使用变性、形成相对链的引物对的退火以及引物延伸的多次循环来呈指数地增加目的核酸序列的拷贝数量。在其变体(也称逆转录pcr(rt-pcr))中,使用逆转录酶(rt)来由mrna制备互补dna(cdna),然后通过pcr来扩增cdna,从而产生大量的dna拷贝。
对于rt-pcr,可使用一步法rt-pcr。一步法rt-pcr是一种能够从rt的孵育至pcr的循环以一个步骤快速方便地进行rt-pcr且无需开闭管或添加试剂的rt-pcr方法。在此技术领域,可使用一步法rt-pcr(比如qiagen的一步法rt-pcrmix)用的各种试剂盒和协议并对其进行适当选择来进行一步法rt-pcr。
pcr的各种其它排列,例如可参见美国专利号4,683,195,4,683,202和4,800,159;mullis等,《酶学方法》,155:335(1987);以及murakawa等,《dna》,7:287(1988);以上各文献的全部内容以引用的方式结合于此。
附图说明
图1示出了核酸扩增装置的构造。
图2示出了pcr芯片的构造。
图3示出了高速实时pcr中的核酸扩增。
图4示出了用于高速实时pcr中的大肠杆菌(escherichiacoli)的校准曲线。
图5示出了对各目的dna长度作图的保留时间,该保留时间允许对目的dna进行充分扩增,即使缩短退火时间和延伸反应时间也没有表现出任何ct值的变化。
图6示出了病原微生物a、病原微生物b和β肌动蛋白基因(βactingene)的多重pcr。
图7示出了在使用不同的逆转录酶的高速一步法逆转录实时pcr中,包含诺如病毒g1或诺如病毒g2基因序列的rna的核酸扩增。
图8示出了在高速一步法逆转录实时pcr中,包括诺如病毒g1或诺如病毒g2基因序列的不同浓度初始的rna的核酸扩增。
图9示出了以循环次数ct(其中在高速一步法逆转录实时pcr中荧光强度迅速增强和上升)对应包含诺如病毒g1或诺如病毒g2基因序列的rna作图得到的校准曲线。
具体实施方式
参照图1-9,本发明反应器的一个实施例解释如下。
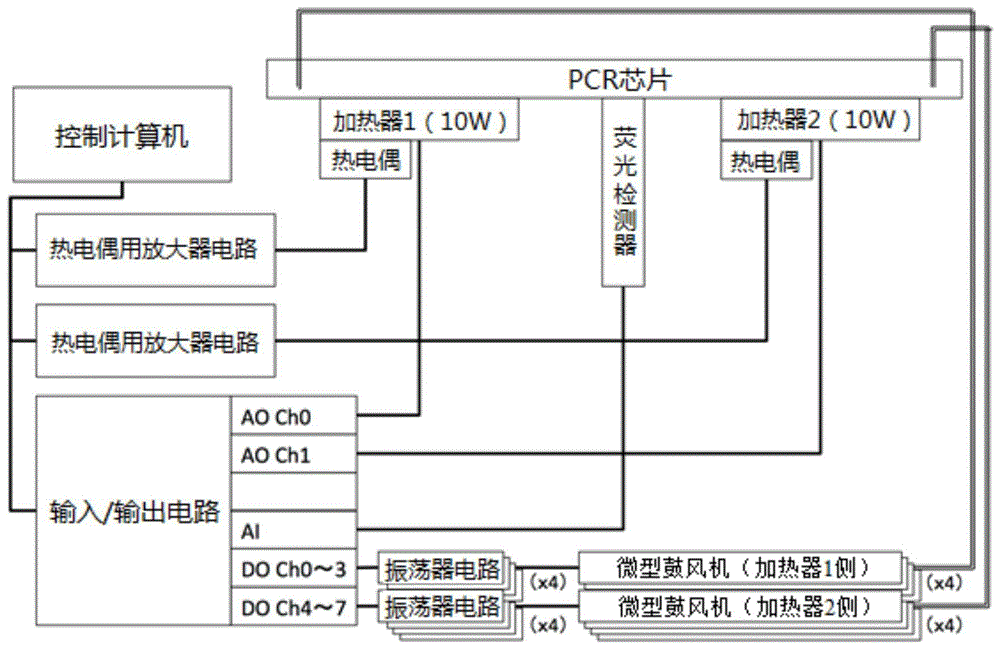
如图1所示,用于高速实时pcr中的装置配置为包括用于在其上放置pcr芯片的基板(图中未示出)、pcr芯片的温度控制部、用作液体输送机构的液体输送微型鼓风机、荧光检测器、用作控制机构的控制计算机、以及供电电池。
pcr芯片的温度控制部配置为包括两个筒形加热器(cartridgeheaters),这两个筒形加热器以10mm的间隔并列设置从而与pcr芯片的蛇形微通道的密封面侧相接触且其间没有任何空隙。为了控制这两个加热器的温度,每个加热器均连接有k型热电偶。
通过控制计算机将筒形加热器1控制为pcr所必需的dna变性反应所需要的温度。该温度(变性温度区)优选为90-100℃,特别优选为95℃。通过控制计算机将筒形加热器2控制为dna的退火反应和延伸反应(延伸/退火温度区)所需的温度。该温度优选为40-75℃,尤其为55-65℃。dna变性反应温度区以及退火反应和延伸反应温度区优选控制在恒定温度。例如,通过pid(比例积分微分)控制使这两个温度区保持在恒定温度。
使用微量吸管之类的将待输送的pcr溶液定量至5-50μl范围之内的所需量,更优选定量至5-25μl。在微量吸管中含有pcr样品溶液状态下,将微量吸管的一次性吸头安装在微通道的一端上。取下微量吸管本体之后,连接上与液体输送微型鼓风机连接的气压管(airpressuretube)作为替代,通过鼓风而施加压力,从而将样品溶液进样至pcr芯片的微通道中。
pcr样品溶液为pcr所需的组分与荧光探针(如taqman探针、cycleave探针或e
荧光检测器设置用于测量位于设置在各微通道中心处的直线微通道上的一个检测点处的荧光强度。当通过施加压力而使从一个蛇形微通通道输送来的pcr溶液流经检测点时,停止液体输送微型鼓风机,从而使pcr溶液可以保留在其它蛇形微通道中一定时间段。
可以对控制计算机进行编程而使其同时控制与各微通道连接的两个微型鼓风机。在对位于各微通道的中心的检测位点处的荧光强度进行连续监测期间,交替地切换微型鼓风机从而将pcr样品溶液交替地移动至各加热器上方的各蛇形微通道一预定时间段,进行热循环。此外,在实时pcr方法中,控制计算机同时记录每次循环的荧光强度的变化和计算循环次数(ct值),其中荧光强度随着目的dna由于实时pcr方法中的热循环的扩增而增加,且荧光强度超过一定的阈值,从而对目的dna的初始量进行定量。
用于高速实时pcr的pcr芯片(核酸扩增用芯片)配置为包括cop树脂基板和施加于基板上的聚烯烃透明密封,该cop树脂基板包括通过注塑形成的四个并列的微通道。
各微通道配置为在两个部分上蜿蜒折回,其宽度如图2所示、其深度为700μm。各蛇形微通道折回四次,并使得位于各微通道中心处的直线微通道夹在多个蛇形微通道之间,且每个蛇形微通道部中可单独容纳至少25μl的溶液。
图2中,使用加热器对虚线围成的区域(变性温度区和延伸/退火温度区)进行加热而进行实时pcr中的热循环。
微通道的两端分别连接至穿透树脂基板的小孔(液体输送机构的连接部)。即使树脂基板的整个微通道侧表面接合有聚烯烃透明密封,反应溶液和空气能通过小孔进入各微通道中。
小孔配置为允许对一般用于生物化学实验中的微量吸管安装一次性的吸头。在量取5-25μl的pcr溶液后,可直接连接一次性吸头,籍此可在无需使用特殊仪器且不受污染等条件下导入pcr溶液。
通过在平坦的基板上形成两个以上微通道或通过并列设置两个以上分别含有微通道的平坦基板,使得中断分析成为可能,从而可以独立地控制通过各微通道的液体输送操作。
优选的,微通道包含的材料具有较高导热性,在pcr所需的温度范围内是稳定的,耐电解质溶液和有机溶剂的腐蚀,并且不吸收核酸或蛋白质。材料的实例包括玻璃、石英、硅和各种塑料。与多个温度区相接触的微通道的形状不仅可以是直线微通道,也可以是弯曲微通道(如环形的蛇形微通道),或螺旋形微通道。微通道的宽度或深度不一定是恒定的,并且微通道可能部分地具有不同的宽度或深度。
优选地,使用相同的荧光检测器进行流经微通道进入不同温度区的样品溶液的通过的检测以及每次热循的环荧光强度的测量,但也可以使用不同的荧光检测器。用于检测样品溶液在不同温度区域之间的通过的方法不限于荧光检测,还可以为光学方法,如比色法和光吸收法,或电法(例如利用电容或包含电化学反应的变化)。与微通道相接触的两个以上温度区可以从微通道的外部与之接触,也可以包含在微通道内。
在图2所示的微通道中,变性温度区和延伸/退火温度区的两个蛇形微通道通过直线中间微通道而彼此连接,在中间微通道中进行样品溶液的通过以及荧光的检测。在图2中,中间微通道沿着连接两个小孔(液体输送机构的连接部)的直线进行设置,并且在中间微通道中进行测试溶液的通过和荧光的检测。因此,即使将pcr芯片(核酸扩增用芯片)翻转(翻转180度)后放置在基板上,也能使用荧光检测器来检测荧光。
例如,如图2所示,pcr芯片可以固定到两个加热器上,其固定方式使得pcr芯片附着在虚线围成的各蛇形微通道部的密封面上,并且使用后,pcr芯片可能被取下、抛弃处理以及替换用于特定用途。两个液体输送微型鼓风机用于pcr芯片的各个微通道。各微型鼓风机分别通过气压管与连接至微通道两端的两个一次性吸头连接。交替地操作各个微型鼓风机,从而实现液体的双向输送。也可以同时对不同的样品进行热循环,例如,根据微通道的数量来增加液体输送微型鼓风机的数量,以进行多重pcr。
本发明上下文中的“多重pcr”是指在单一的反应溶液中使用包含两种以上的正向引物和反向引物的引物集的pcr。本发明上下文中的“引物集”是指一种或两种或更多种的正向引物和反向引物的组合。在本发明中,也可以使用甚至仅包含一种反向引物的引物集作为多重pcr的引物集,只要使用该反向引物与两种以上的正向引物组合(作为引物对)生产出不同的扩增产品即可。
在本发明的多重pcr中,同时测量荧光强度用以检测对应于不同荧光波长的目的基因的扩增。虽然可以使用多个荧光检测器进行检测,即使使用一个波长的光,检测也是可行的。
参照实例,本发明述及如下。然而本发明并不限于这些实例。
实例1:大肠杆菌的定量
使用本发明的高速实时pcr用pcr芯片和装置,对大肠杆菌进行定量。
将大肠杆菌(dh5α株)在卵磷脂肉汤(lecithinbouillon)液体培养基中进行过夜培养。当制备出浓度为1×104cfu/μl的大肠杆菌悬浮液后(基于琼脂平板培养基试验(agarplatemediumassay)的菌落计数(colonycount)),制备一系列10倍稀释液并用作进行定量识别的标准样品。
实时pcr中扩增的目的dna为大肠杆菌-特异uida基因(genbank登录号:nc_000913.3)的一个106bp的dna序列。使用5’-gtgtgatatctacccgcttcgc-3’(序列号:1)作为pcr的正向引物、使用5’-agaacggtttgtggttaatcagga-3’(序列号:2)作为反向引物,pcr溶液中的各引物的最终浓度调整为300nm。实时pcr的
另一种试剂使用日本宝生物(takarabio)公司的
如图3所示,高速实时pcr中每次循环的荧光强度绘制的s形曲线(sigmoidcurve)与由已知实时pcr装置得到的曲线类似。荧光增强率的变化依赖于大肠杆菌的初始浓度。
当跨越荧光强度的期望阈值的循环次数定义为ct,对大肠杆菌的初始浓度绘制校准曲线,得到了良好的线性度,如图4所示。由于非模板对照(notemplatecontrol(ntc),即0cfu/μl)的ct值为45次以上,结果证实,甚至在浓度为100cfu/μl也是可以进行定量。检测灵敏度被证实为与现有的实时pcr装置相当。
高速实时pcr处理时间为每45次循环用时6分钟40秒。相比之下,在现有市售装置中甚至高速热循环装置的pcr处理时间为每45次循环用45分钟。因此,本发明实现了一种能够以极高的速度定量微生物或dna的高速实时pcr。
实例2:高速pcr扩增条件的检查
高速实时pcr的进行重复了三个步骤,即dna变性反应、退火反应和延伸反应;在延伸反应中,使用与dna模板序列对应的dna聚合酶从复制用3’端对各引物进行延伸。其中,dna变性反应和退火反应不依赖于目的dna的长度,并在短时间内完成。然而,延伸反应需要的时间依赖于目的dna的长度和使用的dna聚合酶的酶活性。在热循环高速实时pcr中,适当的时间设置也是必要的。
使用104拷贝数量的大肠杆菌(dh5α株)16s核糖体rna基因(登录号:kc_768803.1)作为模板dna。在得到的模板dna中,在约200-800bp的范围内改变目的dna长度,进行45次循环的高速实时pcr。
使用5’-gtttgatcctggctca-3’(序列号:4)作为通用正向引物序列,使用5’-fam-cgggtgagtaatgtctgg-tamra-3’(序列号:5)作为通用
图5为保留时间对应各目的dna长度绘制的曲线图,该保留时间允许对目的dna进行充足的扩增,而且即使缩短退火时间和延伸反应时间,ct值也没有表现出任何变化。对于约100bp的短目的dna,使用上述uida基因的结果并绘制在同一曲线图中。图5示出了:即使目的dna的长度相同,退火反应时间和延伸反应时间依赖于所使用的dna聚合酶的活性而不同。当使用
理论上,当目的dna的长度为0bp时(对应于图5中的x轴截距),无论使用哪种dna聚合酶,排除延伸反应的退火反应时间约为2.7秒。该结果与前述发现是一致的。因此,从理论上,最高速度的实时pcr可以基于图5并对应于目的dna的长度来设定时间而进行。
实例3:多重pcr高速实时pcr
作为高速实时pcr应用在从同一样品中确认多个目的dna存在与否的多重pcr方法的一个应用实例,使用可同时检测三种荧光的多色荧光检测器同时检测淋病奈瑟菌(neisseriagonorrhoeae)、沙眼衣原体(chlamydiatrachomatis)和人类白细胞衍生β肌动蛋白基因(human-leukocyte-derivedβactingene)。
多色荧光检测器能够对蓝色激发、绿色激发和红色激发的各个荧光进行共轴定量。该检测器可以通过3种荧光探针分别检测荧光增强,即使用fam标记探针、texasred标记探针以及cy5标记探针在pcr芯片上的微通道的同一检测点处进行检测。甚至当使用多色荧光检测器时,检测器的放置方式要使得可以在位于微通道中心处的直线微通道上的同一检测点处同时检测3种荧光强度。当通过施加压力而从一个蛇形微通道部输送来的pcr溶液已通过检测点时,停止液体输送微型鼓风机,从而使pcr溶液可以保留在其它微通道中一定时间段。
将分别用于淋病奈瑟菌、沙眼衣原体和β肌动蛋白基因的目的dna的长度、以及各引物和荧光探针的tm值进行统一,从而避免造成扩增效率的差异。
对于淋病奈瑟菌、沙眼衣原体和β肌动蛋白基因所用的荧光探针,使用texasred标记、cy5标记和fam标记的
pcr溶液中用于淋病奈瑟菌、沙眼衣原体和β肌动蛋白基因的三种正向引物和反向引物各自的最终浓度分别调整为300nm。另一种试剂使用由takarabio公司生产的
制备分别含有各目的dna序列的合成质粒(syntheticplasmids),作为淋病奈瑟菌、沙眼衣原体和β肌动蛋白的模板dna。阳性对照中各质粒的浓度为4ng/μl。而ntc中使用无菌水作为替代进行混合。如此,进行高速实时pcr。热循环条件为:在96℃下加热20秒进行热启动之后,再在96℃下加热3秒,然后在60℃加热8秒,如此重复循环该过程45次。在此条件下,45次循环所用的时间为9分钟40秒。
图6示出了针对淋病奈瑟菌、沙眼衣原体和β肌动蛋白基因使用高速实时pcr的多重pcr结果。由于多色荧光检测器对于三种荧光染料的灵敏度不同,所显示的为通过动态范围校正得到的结果。在图6中,三条粗线示出了当包括所有模板dna时三种荧光的荧光强度的变化。相比于ntc中的荧光信号(由细线表示),得到了清晰的增强并且实现了从同一样品中进行的多项目同时测量。
在本实例中,同时测量三种荧光强度,用以检测对应于各个荧光波长的目的基因的扩增。当通过施加压力从一个蛇形微通道输送来的pcr溶液已经通过检测点后,立即停止液体输送微型鼓风机并进行溶液通过的检测,不需要使用所有的荧光检测器,甚至使用一种波长的光来检测信号,该检测也是可行的。
实例4:一步法逆转录实时pcr
在称之为一步法逆转录实时pcr方法的技术中,使用单一反应溶液方便地进行从rna进行的逆转录反应和实时pcr方法,该单一反应溶液是通过将逆转录酶和pcr溶液进行预先混合制备得到的。该方法已用于检测rna病毒,如流感病毒和诺如病毒。在一步法逆转录实时pcr方法中,将一般rt-pcr方法中的两阶段步骤组合成一个步骤,从而大大简化了操作。然而,这种方法问题在于,逆转录反应的逆转录酶和实时pcr方法的dna聚合酶相互干扰,从而导致pcr效率较差。然而,通过高速温度控制将温度快速转移至逆转录酶和dna聚合酶的活性所需的最佳温度,可以使逆转录反应和实时pcr方法有效地进行,从而完成高效的一步法逆转录实时pcr方法。利用本发明的高速实时pcr用pcr芯片和装置,对采用一步法逆转录实时pcr方法进行的诺如病毒g1基因和g2基因的定量进行了确查。
作为含有目的g1基因或g2基因序列的rna,使用包括在市售takaraqpcr诺如病毒(gi/gii)分型试剂盒(typingkit)中的标准产品、或合成dna的转录产物rna。rna的稀释液系列使用rnase-free无菌水制备进行制备。
使用在日本国家传染病研究所传染病监空中心提供的诺如病毒的检测方法中公开的序列作为引物和探针序列。对于诺如病毒g1基因,其正向引物序列为cog-1f的5’-cgytggatgcgnttycatga-3’(序列号:10);
g1基因和g2基因的荧光探针使用fam标记的
pcr溶液中g1基因或g2基因的正向引物和反向引物的最终浓度分别调整为300nm。对于其它试剂,使用takarabio公司的
热循环条件设置如下。当使用takarabio公司的
高速一步法逆转录实时pcr中的每次循环的荧光强度如图7所示。当诺如病毒g1基因和g2基因的初始浓度相同时,得到不依赖于逆转录酶的类型的类似s形曲线,对于诺如病毒g1基因和g2基因,荧光强度迅速增强和上升时的循环次数是一样的。
其次,高速一步法逆转录实时pcr是通过改变诺如病毒g1基因和g2基因的rna的初始浓度而进行的。如图8所示,荧光强度迅速增强和上升时的循环次数依赖于诺如病毒g1基因的初始浓度而不同。对荧光强度的迅速增强和上升时的循环次数的观察是以rna的初始浓度从最高到最低的顺序进行的。因此,rna的初始浓度一般可通过ct值来定量,其中ct值为荧光强度迅速增强和上升时的循环次数。
然而,如图8所示的诺如病毒g1基因的扩增曲线证实了:基线为一个向上倾斜的曲线,使得荧光强度随着热循环而缓慢增强,然后从一定次数的循环之后荧光强度迅速增加。因此,难以通过常用方法(其中将一定水平的荧光强度定义为阈值,荧光强度高于该阈值时的循环次数定义为ct值)来准确估计ct值。
因此,为了甚至在基线不是恒定值时(即基线成比例增加)检测一步法逆转录实时pcr过程中的ct值,从由各热循环次数确定的荧光强度矩阵(扩增曲线的二维数组)来推导ct值。
为了检测在扩增曲线的二维阵列中相对于ct值以下的斜率迅速上升的斜率,可以进行以下操作。当荧光强度发生较大变化时,如有必要,可求得移动平均。这样做时,是对每次热循环进行第一正向微分。在得到的斜率的新的二维阵列中,对初始阶段的斜率(例如,5-15次循环,或刚好在每次循环之前的5-15次循环)的根均方(可选择性地为加权平均)和初始阶段后的斜率进行比较。当观察到循环次数的显著增加(例如,5倍以上,也可能选择性地为2倍以上)时,由此推断,在该循环次数ct中,荧光强度迅速增强和上升。
图9示出了由得到的ct值对应诺如病毒g1基因和g2基因的初始浓度绘制的校准曲线。相对于诺如病毒g1基因和g2基因的每个rna浓度,获得了良好的线性度。当荧光强度迅速上升时,甚至在一步法逆转录实时pcr中也能快速确定ct值。可以从ct值计算初始rna浓度。
本发明的特征为,系统中全部pcr溶液通过微通道并使得每次热循环中溶液通过荧光检测点。因此,即使由于缺乏时间而不能将荧光染料均匀分散在pcr溶液中(由较多的热循环产生)而致使实时pcr产生的荧光染料在pcr溶液分散不均而形成的荧光染料浓度状态,通过一个荧光检测器也可检测所有的荧光染料,然后对此积算可用来对每次循环的荧光量进行准确定量。
因此,如图9所示,在校准范围内,rna浓度的测量中的各个ct值的误差线是非常小的,并且证实了具有良好的再现性的精确定量是可行的。
工业实用性
根据本发明的装置是便携式的,并且允许在临床情景或在传染病的发生现场以较低的成本进行高速实时pcr。更具体地说,可以通过快速确认治疗效果和尽早地检出家畜和家禽中的传染病来预防感染的传播。
序列表
<110>国立研究开发法人产业技术综合研究所
<120>核酸扩增装置、核酸扩增方法以及核酸扩增用芯片
<130>p15-070
<150>jp2014-140758
<151>2014-07-08
<160>16
<170>patentinversion3.5
<210>1
<211>22
<212>dna
<213>人工序列
<220>
<223>上游引物
<400>1
gtgtgatatctacccgcttcgc22
<210>2
<211>24
<212>dna
<213>人工序列
<220>
<223>下游引物
<400>2
agaacggtttgtggttaatcagga24
<210>3
<211>22
<212>dna
<213>人工序列
<220>
<223>探针
<400>3
tcggcatccggtcagtggcagt22
<210>4
<211>16
<212>dna
<213>人工序列
<220>
<223>上游引物
<400>4
gtttgatcctggctca16
<210>5
<211>18
<212>dna
<213>人工序列
<220>
<223>探针
<400>5
cgggtgagtaatgtctgg18
<210>6
<211>16
<212>dna
<213>人工序列
<220>
<223>下游引物
<400>6
ctttggtcttgcgacg16
<210>7
<211>15
<212>dna
<213>人工序列
<220>
<223>下游引物
<400>7
gcatggctgcatcag15
<210>8
<211>17
<212>dna
<213>人工序列
<220>
<223>下游引物
<400>8
ctgacttaacaaaccgc17
<210>9
<211>18
<212>dna
<213>人工序列
<220>
<223>下游引物
<400>9
taccagggtatctaatcc18
<210>10
<211>20
<212>dna
<213>人工序列
<220>
<223>上游引物
<220>
<221>misc_feature
<222>(12)..(12)
<223>nisa,c,g,ort
<400>10
cgytggatgcgnttycatga20
<210>11
<211>20
<212>dna
<213>人工序列
<220>
<223>探针
<400>11
agatygcgatcycctgtcca20
<210>12
<211>20
<212>dna
<213>人工序列
<220>
<223>探针
<400>12
agatcgcggtctcctgtcca20
<210>13
<211>21
<212>dna
<213>人工序列
<220>
<223>下游引物
<400>13
cttagacgccatcatcattya21
<210>14
<211>26
<212>dna
<213>人工序列
<220>
<223>上游引物
<220>
<221>misc_feature
<222>(9)..(9)
<223>nisa,c,g,ort
<400>14
cargarbcnatgttyagrtggatgag26
<210>15
<211>20
<212>dna
<213>人工序列
<220>
<223>探针
<400>15
tgggagggsgatcgcratct20
<210>16
<211>21
<212>dna
<213>人工序列
<220>
<223>下游引物
<400>16
tcgacgccatcttcattcaca21
- 还没有人留言评论。精彩留言会获得点赞!
- 一种基于CRISPR‑Cas9系统的基因编辑载体及其应用的制造方法与工艺
- 一种基于CRISPR/Cas9技术制备水稻OsPIL15突变体的方法及应用与流程
- 检测黄曲霉毒素M1的核酸适配体、传感器、试剂盒及应用的制造方法与工艺
- 一组肌酸激酶同工酶核酸适配体及其应用的制造方法与工艺
- 用于抑制KRAS靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 用于抑制EIF4E靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 用于抑制VEGFA靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 一种新型向导RNA表达盒及在CRISPR/Cas系统中的应用的制造方法与工艺
- 一种具备双单链末端PCR产物的获得方法及其检测方法与流程
- 一种通过生物技术扩增脱氧胸苷三磷酸的方法与流程