一种多黏菌素E组分及其光化学产物和液相色谱-质谱分析方法与流程
本发明属于药物化学和药物分析化学领域,尤其涉及一种多黏菌素e(polymyxine,pme)组分及其光化学产物和液相色谱-质谱分析方法。
背景技术:
多黏菌素e(pme)由多黏类芽孢杆菌发酵产生的多组分脂肽类混合物,临床上主要应用硫酸pme(或称之为黏菌素,colistin)或多黏菌素e甲磺酸钠作为“最后一道防线”来治疗多重耐药革兰阴性菌感染。由于单独使用pme时需提高剂量,但肾毒性随之增加,因而采用联合用药或雾化吸入成为一种减小毒性、提高治疗效果的趋势,另外,开发低毒、高效的pme单一组分也成为近年来的研究热点(jmedchem,2018,61(5):1845-1857)。
pme主要组分(e1、e2、e3、e1-i和e1-7'moa)的结构通式及信息如下:
pme主要组分由n-端脂肪酰基链(fattyacyl,fa)、线性三肽及环状七肽构成,1、3、4、8及9位均为l-α,γ-二氨基丁酸(dab),2及10位为l-苏氨酸(thr),6位为d-亮氨酸(leu),7位为l-leu或异亮氨酸(ile)。不同组分的脂肪链取代基或氨基酸的不同造成结构之间存在差异,对多黏菌素组分的分析方法主要有液相色谱(《欧洲药典》9.0版)、液质联用(jchromatoa,2002,976(2):65–78)及毛细管电泳方法(electrophoresis,2000,21(18):3199-3204)。由于pme是含有多种组分的混合物,对其组分分析及结构鉴定具有一定的难度。虽然目前已有二十余种pme组分被发现,但由于不同的发酵菌或发酵条件而产生出其他不同组分,该类组分有着不同的抗菌活性及毒性。目前仍存在未被发现的pme组分。
技术实现要素:
为了克服现有技术存在的问题,本发明公开了一类新的多黏菌素e双键化组分,具体为多黏菌素e双键化组分7'-烯基多黏菌素e1和6'-烯基多黏菌素e2的结构,并首次经hplc-ms分析方法结合二级质谱确证,并经光化学反应即帕特诺-比希反应(
为实现上述目的,本发明采用如下技术方案:
本发明的第一个方面是提供一种多黏菌素e组分,其包括多黏菌素e双键化组分。
进一步地,所述多黏菌素e双键化组分包括多黏菌素e1双键化组分和多黏菌素e2双键化组分。
进一步地,所述多黏菌素e1双键化组分为4'-烯基多黏菌素e1、5'-烯基多黏菌素e1、6'-烯基多黏菌素e1或7'-烯基多黏菌素e1;所述多黏菌素e2双键化组分为4'-烯基多黏菌素e2、5'-烯基多黏菌素e2或6'-烯基多黏菌素e2。
进一步地,所述双键位于n-脂肪酰基链末端,所述多黏菌素e1双键化组分为7'-烯基多黏菌素e1,所述多黏菌素e2双键化组分为6'-烯基多黏菌素e2;
其中,所述7'-烯基多黏菌素e1的结构式如下:
其中,所述6'-烯基多黏菌素e2的结构式如下:
其中,上述双键的位置经hplc-ms及二级质谱分析验证位于n-脂肪酰基链末端。
进一步地,所述多粘菌素e组分来源于多黏菌素e原料药、硫酸多黏菌素e制剂、磺酸多黏菌素e制剂或多黏菌素e甲磺酸钠制剂,其也可来源于其他制剂。
进一步地,所述多黏菌素e组分的组成单元包括n-脂肪酰基链、α,γ-二氨基丁酸、苏氨酸、亮氨酸或异亮氨酸、缬氨酸或正缬氨酸、丝氨酸、甲硫氨酸,也可包括其他氨基酸。
进一步地,氨基酸为l-构型或d-构型。
进一步地,所述n-脂肪酰基链包括6'-甲基辛酰基(pme1的n-脂肪酰基)、6'-甲基庚酰基(pme2的n-脂肪酰基)、7'-甲基辛酰基(e1-7'moa的n-脂肪酰基)、辛酰基(pme3的n-脂肪酰基)、庚酰基(pme4的n-脂肪酰基)、3'-羟基6'-甲基辛酰基(pme6的n-脂肪酰基)、3'-羟基6'-甲基庚酰基、己酰基,也可包括其他n-脂肪酰基。
进一步地,所述n-脂肪酰基链的末端均可双键化。
进一步地,所述多黏菌素e主要组分为多黏菌素e1和多黏菌素e2,其他组分包括多黏菌素e3、多黏菌素e4、多黏菌素e6、多黏菌素e1-7'moa、多黏菌素e1-i、多黏菌素e2-i、多黏菌素e1-val、多黏菌素e2-val、多黏菌素e1-ser、多黏菌素e2-ser,也可包括另外的其他组分。
进一步地,所述多黏菌素e组分中的双键化组分包括多黏菌素e氧化产物、差向异构化产物、水解产物、硫酸化产物、磺酸化产物、甲磺酸化产物,也可为其他产物。
本发明的第二个方面是提供一种任一上述的多黏菌素e组分的光化学产物,所述多黏菌素e组分与羰基化合物通过光化学反应得到[2+2]环加成产物。
进一步地,所述羰基化合物包括丙酮、丁酮、2-戊酮、3-戊酮、己酮、苯甲酮、苯乙酮、甲醛、乙醛、丙醛、丁醛、戊醛,也可为其他形式的羰基化合物,优选为丙酮。
进一步地,所述多黏菌素e组分为7'-烯基多黏菌素e1,所述环加成产物包括羰基氧与7'-碳加成、或羰基氧与8'-碳加成两种区域选择性产物,加成产物7'-碳的手性是r型或s型。
进一步地,所述7'-烯基多黏菌素e1的环加成产物的结构式为
进一步地,所述多黏菌素e组分为6'-烯基多黏菌素e2,所述环加成产物包括羰基氧与6'-碳加成、或羰基氧与7'-碳加成两种区域选择性产物,加成产物6'-碳的手性是r型或s型。
进一步地,所述光化学反应采用的溶剂为水、水:乙腈或水:甲醇;优选为水:乙腈=3~5:1,更优选为水:乙腈=4:1。
进一步地,在所述光化学反应中,所述羰基化合物为丙酮,所述溶剂与丙酮的体积比为1:9-9:1,优选为1:4-4:1,更优选为1:1。
本发明的第三个方面是提供一种用于分析多黏菌素组分或多肽类抗生素组分的液相色谱-质谱分析方法,其采用高分辨一级质谱、二级质谱碎片离子及氨基酸残基离子信息推断组分结构。
进一步地,所述多黏菌素组分为多黏菌素e1双键化组分和多黏菌素e2双键化组分;其中,所述多黏菌素e1双键化组分为4'-烯基多黏菌素e1、5'-烯基多黏菌素e1、6'-烯基多黏菌素e14或7'-烯基多黏菌素e1;所述多黏菌素e2双键化组分为4'-烯基多黏菌素e2、5'-烯基多黏菌素e2或6'-烯基多黏菌素e2。
进一步地,所述液相色谱选用反相色谱柱,填料为十八烷基键合的硅胶基质、八烷基键合的硅胶基质或四烷基键合的硅胶基质,色谱柱柱长5-30cm,直径1-10mm,填料粒径1-10μm;优选为c18色谱柱,25cm,4.6mm,5μm。
进一步地,所述多黏菌素e的终浓度为0.1mg/ml-20mg/ml优选为0.5~4mg/ml,更优选为2mg/ml。
进一步地,所述液相色谱方法中,流动相a为含三氟乙酸水溶液:乙腈,其体积比为99:1-80:20,三氟乙酸含量为0.01%-1%(v:v);流动相b为乙酸、甲醇、或含甲酸的乙腈溶液,甲酸含量为0.01%-1%(v:v)。优选流动相a为含0.1%(v:v)三氟乙酸水溶液:乙腈=95:5,流动相b为含0.1%(v:v)甲酸的乙腈溶液。
进一步地,所述液相色谱方法中,采用等度洗脱,流动相a与流动相b的洗脱体积比例为70:30-95:5;优选为80:20。
进一步地,所述液相色谱方法中,流速为0.5-1.5ml/min;优选为1ml/min。
进一步地,所述液相色谱方法中,进样体积为1-100μl;优选为4μl。
进一步地,所述液相色谱方法中,色谱柱柱温选择在20℃-40℃;优选为30℃。
进一步地,质谱离子化方式选择电喷雾离子化、大气压离子化或快原子轰击离子化,也为其他离子化方式;优选为电喷雾离子化,正离子采集模式。
进一步地,质谱的质量分析器选择四级杆飞行时间质谱、三重四级杆质谱、离子肼质谱或轨道离子肼质谱,也可为其他质量分析器;优选为四级杆飞行时间质谱。
进一步地,质谱及质谱/质谱扫描范围为m/z50-1700。
进一步地,二级质谱碰撞能量设置范围为5-50ev;优选为20ev。
进一步地,所述7'-烯基多黏菌素e1的分子量为1166.75,双键组分的特征离子为m/z239.17、139.11、111.11、69.07和55.05。
进一步地,所述6'-烯基多黏菌素e2的分子量为1152.74,双键组分的特征离子为m/z225.16、125.10、97.10和55.05。
进一步地,所述7'-烯基多黏菌素e1光化学产物的分子量为1224.80,环氧化产物的特征离子为m/z899.56、799.50、498.33、297.22、171.11和157.10。
本发明的第四个方面是提供一种药物中含有双键结构的杂质鉴定方法及应用,其采用药物与任一上述的羰基化合物进行光化学反应,并通过液相色谱-质谱分析方法进行分析。
与现有技术相比,本发明采用上述技术方案具有以下有益效果:
(1)本发明首次发现多黏菌素e中含有组分6'-烯基pme2,并首次公布了7'-烯基pme1和6'-烯基pme2的结构;
(2)本发明首次通过光化学反应即帕特诺-比希反应获得了7'-烯基pme1的环氧化产物;
(3)本发明中的hplc-ms方法可直接分析多黏菌素e中的组分,总结了组分结构分析的通用策略,即综合采用高分辨一级质谱、二级质谱碎片离子及氨基酸残基离子信息推断组分结构,简单快捷,并可推广至其他多肽类抗生素的结构解析中;
(4)本发明中的hplc-ms方法包含了一条新的裂解途径可帮助辅助判断多黏菌素e的组分结构;
(5)本发明中的双键化组分及其光化学反应产物的结构解析策略适合于其他多黏菌素双键化组分或多肽类抗生素双键化组分,易于推广。
本发明建立了pme的hplc-ms的组分分析方法,确立了pme组分结构解析的通用策略,即综合采用高分辨一级质谱、二级质谱碎片离子及氨基酸残基离子信息推断组分结构,并发现了一条新的裂解途径。另外,获得了pme中双键化组分的光化学反应产物,结合结构解析策略,推断双键化组分结构为7'-烯基多黏菌素e1,并首次发现了6'-烯基多黏菌素e2。本发明所涉及的结构解析策略也可应用至其他脂肽类化合物的结构推断中,光化学反应也为药物中含有双键结构的杂质鉴定提供了一种新颖的解决思路,对产品工艺改进及质量控制水平提高具有一定的指导意义。
附图说明
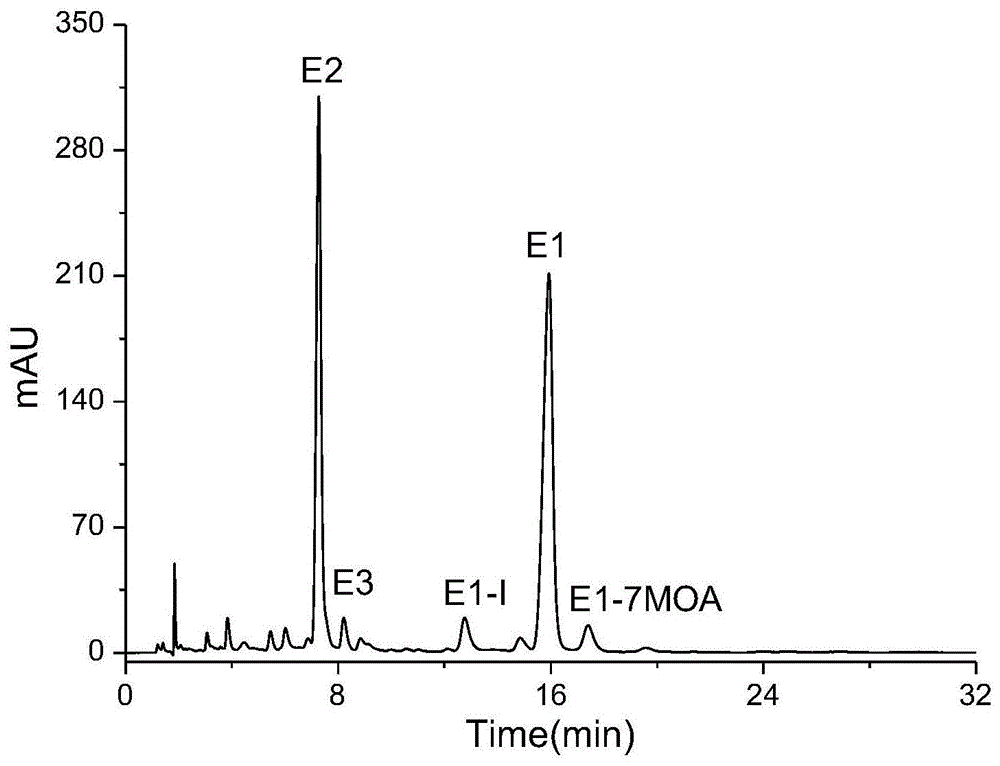
图1为本发明一实施例中多黏菌素e对照品(《欧洲药典》对照品)通过《欧洲药典》9.0版中的分析方法获得的典型色谱图。
图2为本发明一实施例中分析多黏菌素e对照品的质谱总离子流图,图中峰14为7'-烯基多黏菌素e1,峰8'为6'-烯基多黏菌素e2。
图3为本发明一实施例中多黏菌素e1的一级质谱及二级质谱图。
图4为本发明一实施例中多黏菌素e1的二级质谱的裂解规律。
图5为本发明一实施例中7'-烯基多黏菌素e1的二级质谱图。
图6为本发明一实施例中6'-烯基多黏菌素e2的二级质谱图。
图7为本发明一实施例中7'-烯基多黏菌素e1光化学产物的二级质谱图。
具体实施方式
本发明涉及一类新的多黏菌素e组分及其光化学产物和hplc-ms分析方法。所述组分中包括多黏菌素e1双键化组分、多黏菌素e2双键化组分,该类组分经hplc-ms及二级质谱分析验证双键位于n-脂肪酰基链末端。所述双键化组分在丙酮做反应试剂的条件下经光化学反应即帕特诺比希反应获得光化学产物,该类产物经hplc-ms验证为[2+2]环加成产物,上述双键化组分被命名为7'-烯基多黏菌素e1和6'-烯基多黏菌素e2。
下面结合附图和实施例,对本发明的具体实施方式作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
下述实施例中的多黏菌素e,以硫酸多黏菌素e《欧洲药典》对照品为具体实施案例,但也可适用于其他多黏菌素e对照品及制剂中。
实施例1
一、仪器与试药
agilent1260高效液相色谱仪、agilent1290型高效液相色谱仪-6550qtof-ms(美国agilenttechnologies公司),wfh-203b三用紫外分析仪(上海驰唐电子有限公司)。
硫酸多黏菌素e《欧洲药典》对照品(批号:3.3)。
二、实验条件
按照《欧洲药典》9.0版中多黏菌素e的组分分析方法,选择diamonsilplusc18色谱柱(250mm×4.6mm,5μm),以硫酸钠溶液:乙腈为流动相,流速为1ml/min;柱温为30℃;进样量为20μl;检测波长为215nm。
本实施例中以含0.1%三氟乙酸水溶液:乙腈=95:5为流动相a,含0.1%甲酸的乙腈溶液为流动相b梯度洗脱,流速为1ml/min;柱温为30℃;进样量为4μl。以esi-q/tof-ms为检测器,采用正离子扫描,采集一级质谱图和二级质谱图,采集范围m/z50-1700,二级质谱碰撞能量为20ev。
三、多黏菌素e对照品样品溶液配制及分析
采用《欧洲药典》9.0版中的分析方法:精密称取多黏菌素e对照品10mg,加水-乙腈(80:20)5ml溶解,浓度为2mg/ml。经hplc-uv分析,其典型色谱图见图1所示,通过相对保留时间对多黏菌素e1、e2、e3、e1-i和e1-7'moa进行定位。
采用本实施例中的方法:以上多黏菌素e对照品溶液采用本实施例中实验条件进行分析,总离子流图见图2所示,经一级质谱及二级质谱确认,图中峰15为多黏菌素e1,峰8为多黏菌素e2,峰9为多黏菌素e3,峰12为多黏菌素e1-i,峰17为多黏菌素e1-7’moa,峰14为7'-烯基多黏菌素e1,峰8'为6'-烯基多黏菌素e2。
四、多黏菌素e1的结构解析
以多黏菌素e1为例介绍结构解析过程,其一级质谱为多电荷峰,准分子离子峰[m+h]+的m/z为1169.77,[m+2h]2+为585.39,[m+3h]3+为390.5964,也可以观察到[m+na]+(m/z,1191.75),根据准分子离子峰、多电荷峰以及加合金属离子峰所可以帮助确认多黏菌素组分的精确分子量,即e1的分子量为1168.77。多黏菌素e1的[m+2h]2+相对丰度最高,以其为母离子,在20ev下可以得到丰富的二级质谱碎片信息,得到的多黏菌素e1二级质谱图如图3所示,碎片离子的归属见图4所示。e1的第一系列碎片离子(见图3中74.1、227.2、327.2、427.3、527.3、728.5、829.5和929.6以及图4中最后两行)首先失去6-甲基辛酰基和α,γ-二氨基丁酰基、苏氨酰基得到环肽碎片离子([m+h]+,728.48),继续逐一失去α,γ-二氨基丁酰基、苏氨酰基,最后得到只有d-leu和l-leu的二肽碎片离子;第二系列碎片离子丰度较低(见图7中297.22、498.33、799.50和899.56),多黏菌素e1首先丢失环肽上的d-leu和l-leu得到碎片离子m/z843.54,继而从环肽端逐渐丢失α,γ-二氨基丁酰基、苏氨酰基等,最后得到6-甲基辛酰基的碎片离子。图3中的101.1、202.1、302.2、402.2、503.3和603.4与图4中的603.36、503.29、402.25、302.18、202.12、101.07和703.42展示了多黏菌素e的一条新的裂解途径,得到的碎片离子称之为第三系列离子,碎片离子m/z843.54也会从脂肽端首先失去n-端脂肪酰基,继而丢失α,γ-二氨基丁酰基、苏氨酰基等得到一系列小肽离子,该系列离子可以辅助判断多黏菌素e某些位点上的氨基酸取代。低端分子量区域显示了组成多黏菌素e的氨基酸的特征离子,如leu或ile的特征离子m/z86.10,dab的为m/z101.07和73.08,thr的为m/z84.04(β位羟基发生脱水反应)和74.06。如果低端分子量发生了变化,则可直接反映出多黏菌素的氨基酸的组成发生了变化。
五、7'-烯基多黏菌素e1和6'-烯基多黏菌素e2的结构解析
峰14中组分即7'-烯基多黏菌素e1的第一系列及第三系列碎片离子显示与多黏菌素e1基本一致的信号,见图5;而第二系列碎片离子的m/z均比多黏菌素e1的小2,特征离子m/z239.17、139.11、111.11、69.07和55.05证实双键位于脂肪链末端。峰8'中的组分即6'-烯基多黏菌素e2其[m+2h]2+为577.37,分子量比e2的少2;ms/ms中第一系列离子同e2的基本一致,第二系列离子高质量端的离子丰度较小,但低质量端m/z225.16和125.10说明双键在脂肪链端,特征离子m/z97.10和55.05证明双键发生在末端,二级质谱图见图6。
五、7'-烯基多黏菌素e1光化学产物的结构解析
7'-烯基多黏菌素e1光化学产物的二级质谱图见图7。第一系列离子与多黏菌素e1的基本一致,第二系列离子中m/z899.56、799.50、498.33、297.22证明与丙酮的加成反应。在谱图中发现新的特征离子m/z171.11和157.10,有两种可能产生路径,其一是丙酮上的羰基与7'位碳结合(不考虑7'碳的手性),氧孤对电子促进7'位碳上的h发生[1,5h]或[1,6h]转移分别生成m/z171.11和157.10,其二是丙酮上的羰基与8'位碳结合(不考虑7'碳的手性),氧孤对电子促进8'位碳上的h发生[1,6h]或[1,7h]转移分别生成m/z171.11和157.10。该实验及数据证明双键位于n-脂肪链末端。
由上述实施例可知,本发明中发现了新的多黏菌素e双键化组分,并可通过hplc-ms方法解析其结构。光化学反应可以得到双键化组分的环氧化产物,并通过hplc-ms方法解析了结构,验证了双键位于n-脂肪链末端。本发明提出了多黏菌素组分通用的解析策略和证明双键化组分的方法,可适用于其他多肽类抗生素尤其是双键化组分的结构鉴定中。
以上对本发明的具体实施例进行了详细描述,但其只作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!