一种胺脱氢酶及其应用的制作方法
本发明属于基因工程技术领域,具体涉及一种胺脱氢酶及其应用。
背景技术:
手性胺是许多生物活性分子的结构单元,是重要的医药和精细化工手性砌块。约40%的光学活性药物含有手性胺结构。手性胺高效制备技术是手性药物领域发展的关键技术。现有技术中,手性胺的化学制备方法主要包括氨基酸或其衍生物的还原、醛或酮与亚胺的偶合以及环氧化合物的氨基化开环等,这些化学制备方法的催化效率较低且容易造成环境污染。因此,高效生物催化是解决手性胺制造并减少环境污染等问题的重要途径。生物法制备手性胺主要通过酶催化拆分或不对称还原实现,现有技术中用于制备手性胺的酶主要包括脂肪酶、转胺酶和氧化还原酶。而胺脱氢酶催化酮和氨基供体不对称还原胺化制备手性胺简洁高效而且易实现辅酶再生。
苯丙氨酸脱氢酶催化天然氨基酸苯丙氨酸的氧化脱氨或还原氨化。然而对于系列酮的还原胺化制备手性胺的基本没有催化活性。通过定点突变对野生型酶进行改造,获得胺脱氢酶,提高其催化还原胺化的活性。
技术实现要素:
本发明的目的在于提供一种胺脱氢酶。
本发明的另一目的在于提供上述胺脱氢酶的应用。
本发明的技术方案如下:
一种胺脱氢酶,对如seqidno.01所示的氨基酸脱氢酶进行突变而获得,上述突变包括如下至少之一:
将第131位的g突变为l或m、将第262位的n突变为v或l、将第285位的y突变为l或m和将第333位的m突变为d。
在本发明的一个优选实施方案中,所述突变为将第131位的g突变为l或m、将第262位的n突变为v或l、将第285位的y突变为l或m或将第333位的m突变为d。
在本发明的一个优选实施方案中,所述氨基酸脱氢酶的核苷酸序列如seqidno.02所示。
进一步优选的,所述突变通过pcr进行,其中,将第131位的g突变为l所用的引物为g131l-f和g131l-r,其核苷酸序列分别如seqidno.03和04所示,将第131位的g突变为m所用的引物为g131m-f和g131m-r,其核苷酸序列分别如seqidno.05和06所示。
进一步优选的,所述突变通过pcr进行,其中将第262位的n突变为v所用的引物为n262v-f和n262v-r,其核苷酸序列分别如seqidno.07和08所示,将第262位的n突变为l所用的引物为n262l-f和n262l-r,其核苷酸序列分别如seqidno.09和10所示。
进一步优选的,所述突变通过pcr进行,其中将第285位的y突变为l所用的引物为y285l-f和y285l-r,其核苷酸序列分别如seqidno.11和12所示,将第285位的y突变为m所用的引物为y285m-f和y285m-r,其核苷酸序列分别如seqidno.13和14所示。
进一步优选的,所述突变通过pcr进行,其中将第333位的m突变为d所用的引物为m333d-f和m333d-r,其核苷酸序列分别如seqidno.15和16所示。
本发明的另一技术方案如下:
上述胺脱氢酶在催化不对称还原胺化得到手性胺中的应用。
该应用的反应式如下:
在本发明的一个优选实施方案中,构建反应体系,于10-70℃且100-300rpm的转速震荡反应10-90h,获得所述手性胺;
上述反应体系包括如下:ph=8-12,0.001-0.05mol/l酮,0.002-0.2mol/l氨基供体,0-0.2mol/l辅助底物,0.01-0.2mol/l的缓冲液,5-300mg/l的所述胺脱氢酶;
上述酮的结构式为
进一步优选的,所述氨基供体为丙氨酸、氨水、nh4cl和nh4no3中的至少一种,所述缓冲液为tris-盐酸缓冲液、乙酸钠缓冲液、碳酸钠缓冲液和磷酸钾缓冲液中的至少一种,所述辅助底物为nadh。
本发明的有益效果是:
1、本发明通过氨基酸脱氢酶突变获得的新型胺脱氢酶,对r型的选择性更好;且对底物酮催化偏好与现有的酶不同,其对芳香酮以及分子式为cxhyo(其中x为4-7,y为8-14,酮的分子量为72-115)的脂肪族酮具有较好的催化活性和选择性;该胺脱氢酶的获得及其催化特性的挖掘拓展了不对称还原制备手性胺的范围。
2、本发明的胺脱氢酶的催化反应操作方便,具有产品光学纯度高、收率高等优点,设备简单,在生物催化制备手性胺和海洋生物资源高值化开发领域具有较好的工业应用前景。
附图说明
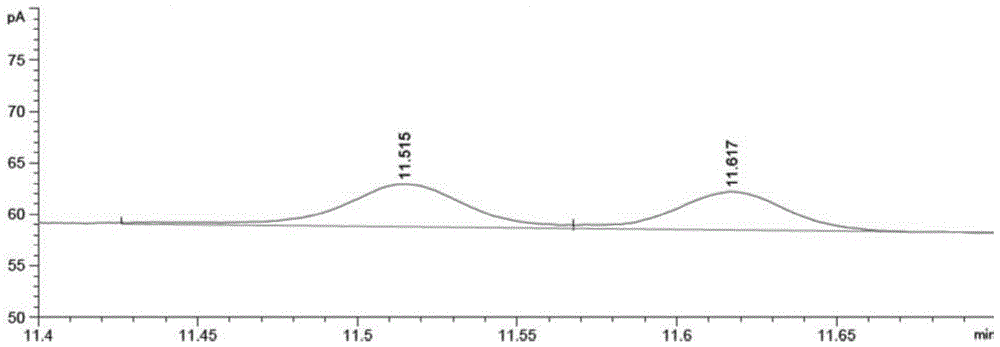
图1为本发明实施例1中涉及的1,3-二甲基丁胺标准样品衍生后色谱图。(s)-1,3-二甲基丁胺和(r)-1,3-二甲基丁胺的保留时间分别为11.515min和11.617min。
图2为本发明实施例2中涉及的5-甲基-2-己酮、5-甲基-2-己胺色谱图。底物5-甲基-2-己酮的保留时间为8.658min,(s)-5-甲基-2-己胺和(r)-5-甲基-2-己胺的保留时间分别为14.529min和14.665min。
具体实施方式
以下通过具体实施方式结合附图对本发明的技术方案进行进一步的说明和描述。
实施例1
构建单点突变(g131l)的氨基酸脱氢酶突变体,对131位氨基酸进行突变,具体步骤如下:
(1)引入突变:根据seqidno.02所示的核苷酸序列设计引物,设计包含有不同位点的上游引物和下游引物,所述上游引物和下游引物如下:
上游引物(seqidno.03):g131l-faattgtatcgtcctcgtacccgaa
下游引物(seqidno.04):g131l-rgaggacgatacaattcgtttcttt
将引物和模板质粒混合后,加入高保真taq聚合酶kod-plus,进行全质粒pcr扩增,pcr结束后电泳检测pcr产物。
(2)构建可表达带his-tag标签的苯丙氨酸脱氢酶的重组表达菌株:苯丙氨酸脱氢酶(phedh)基因的原始序列来自bacillusnanhaiensis,如seqidno.02所示,原始氨基酸序列如seqidno.01所示;根据上述原始序列,设计如seqidno.03和seqidno.04所示的引物,采用pcr法构建5’端和3’端分别带有ndei和xhol酶切位点的苯丙氨酸脱氢酶基因,pcr合成过程由上海生工生物工程技术服务有限公司完成。pcr扩增产物经1%琼脂糖凝胶电泳鉴定后,胶回收phedh基因片段,用ndei和xhoi酶切酶进行双酶切,回收酶切产物,与同样双酶切的pet-28a质粒(带有his-tag标签)进行连接,连接好的质粒转化到大肠杆菌bl21(de3),得到pet28a-phedh质粒。将上述质粒转化至e.colibl21(de3),得到可表达带his-tag标签的胺脱氢酶的重组表达菌株e.colibl21(de3)/pet28a。
(3)重组大肠杆菌e.colibl21(de3)/pet28a的培养:以1%的接种量,将菌种接入200mllb培养基中。lb培养基的组成为10.0g/l胰蛋白胨,5.0g/l酵母浸膏,10g/lnacl。培养条件为:起始ph7.0,装液量体积分数为10%,培养温度37℃,摇床转速200rpm,培养时间6h。加入诱导剂iptg,使其终浓度为10mg/ml,继续在25℃、200rpm条件下培养12h。
(4)粗酶液的制备:培养结束获得的发酵液,在冷冻离心机中离心(4℃,8000rpm,15min)获得细胞,弃上清液,沉淀用磷酸缓冲液(ph=7)重悬,充分洗涤后离心,重复操作3次,用磷酸缓冲液(ph7)配制成浓度为50g/l的细胞悬液。将制备的细胞悬液置于冰浴中,利用超声破碎仪对细胞液进行处理,细胞破碎仪探头置于液面下1cm,破碎条件为超声3秒,间隔6秒,超声60次,功率200w。然后4℃、12,000rpm离心15min去除不溶性细胞碎片,上清即为含有带his-tag标签的胺脱氢酶的粗酶液。
(5)胺脱氢酶纯酶制备:采用ge公司的histrap镍柱(histraptmhp,5ml)对步骤(3)得到的粗酶液进行分离纯化,并用pall公司的10k的超滤离心管进行超滤除盐。所述的纯化过程采用的纯化柱为能够特异性纯化带有his-tagged标签的蛋白质的histraphp柱,其步骤包括平衡、上样、平衡、洗脱、柱子再生;收集洗脱的部分并利用超滤离心管进行除盐;除盐后获得的液体即为纯化的带his-tag标签的胺脱氢酶的纯酶溶液。
(6)酶催化:胺脱氢酶的活性测定反应体系包含40mmnadh,50mm4-甲基-2-戊酮,0.2mol/lnh4cl,50mmnadh,0.05mol/l的乙酸钠缓冲液,50mg/l的酶,反应体系ph11,在反应温度20℃下,200rpm震荡反应,反应时间45h。
(7)分离与检测:反应结束后将反应液加入50ml离心管,加入1ml10mnaoh溶液终止反应,再加入3.6gnacl粉末和2ml甲基叔丁基醚,用振荡器混匀,再用摇床300rpm震荡20min,在10000rpm下离心10min。取上层有机相。用2ml甲基叔丁基醚重复萃取一次,混合两次萃取液。在有机相中加入50μl三氟乙酸酐在瓶口包上封口膜,微波炉中火加热3min,取出用甲基叔丁基醚定容至2ml。
将衍生后的样品配合1,3-二甲基丁胺标准样品,用agilenttechnologies7890agcsystem进行气相色谱分析,进样量1μl,气相色谱检测条件:色谱柱型号:安捷伦cyclosil-b手性柱。前进样口:温度250℃,压力11.258psi,流速25ml/min,隔垫吹扫流量3.0ml/min,色谱柱流量:1ml/min,前检测器:温度250℃,燃气流量40.00ml/min,尾吹气流量:50ml/min。最佳升温程序为:初温50℃,每分钟升温5℃,升至80℃再以每分钟升温10℃,升至120℃保持2min。在该色谱条件下对底物和产物进行测定,结果如图1所示。
收率的定义为反应结束后得到产物的摩尔数和反应开始时加入底物的摩尔数的比值;产品胺的对映体纯度的计算公式为:e.e.%=(r-s)/(r+s)×100%,其中s代表s-胺的含量,r代表r-胺的含量。得到的产物为1,3-二甲基丁胺,计算产物胺的收率为93.5%,e.e.值为94.9%。
实施例2
构建单点突变(g131m)的氨基酸脱氢酶突变体,对131位氨基酸进行突变,具体步骤如下:
(1)引入突变:根据seqidno.02所示的核苷酸设计引物设计包含有不同位点的上游引物和下游引物,所述上游引物和下游引物如下:
上游引物(seqidno.05):g131m-faattgtatcgtcatggtacccgaa
下游引物(seqidno.06):g131m-rcatgacgatacaattcgtttcttt
将引物和模板质粒混合后,加入高保真taq聚合酶kod-plus,进行全质粒pcr扩增,pcr结束后电泳检测pcr产物。
(2)构建可表达带his-tag标签的苯丙氨酸脱氢酶的重组表达菌株:苯丙氨酸脱氢酶(phedh)基因的原始序列来自bacillusnanhaiensis,如seqidno.02所示,原始氨基酸序列如seqidno.01所示;根据上述原始序列,设计如seqidno.05和seqidno.06所示的引物,采用pcr法构建5’端和3’端分别带有ndei和xhol酶切位点的苯丙氨酸脱氢酶基因,pcr合成过程由上海生工生物工程技术服务有限公司完成。pcr扩增产物经1%琼脂糖凝胶电泳鉴定后,胶回收phcdh基因片段,用ndei和xhoi酶切酶进行双酶切,回收酶切产物,与同样双酶切的pet-28a质粒(带有his-tag标签)进行连接,连接好的质粒转化到大肠杆菌bl21(de3),得到pet28a-phedh质粒。将上述质粒转化至e.colibl21(de3),得到可表达带his-tag标签的胺脱氢酶的重组表达菌株e.colibl21(de3)/pet28a。
(3)重组大肠杆菌e.colibl21(de3)/pet28a的培养:以1%的接种量,将菌种接入200mllb培养基中。lb培养基的组成为10.0g/l胰蛋白胨,5.0g/l酵母浸膏,10g/lnacl。培养条件为:起始ph7.0,装液量体积分数为10%,培养温度37℃,摇床转速200rpm,培养时间6h。加入诱导剂iptg,使其终浓度为10mg/ml,继续在25℃、200rpm条件下培养12h。
(4)粗酶液的制备:培养结束获得的发酵液,在冷冻离心机中离心(4℃,8000rpm,15min)获得细胞,弃上清液,沉淀用磷酸缓冲液(ph=7)重悬,充分洗涤后离心,重复操作3次,用磷酸缓冲液(ph7)配制成浓度为50g/l的细胞悬液。将制备的细胞悬液置于冰浴中,利用超声破碎仪对细胞液进行处理,细胞破碎仪探头置于液面下1cm,破碎条件为超声3秒,间隔6秒,超声60次,功率200w。然后4℃、12,000rpm离心15min去除不溶性细胞碎片,上清即为含有带his-tag标签的胺脱氢酶的粗酶液。
(5)胺脱氢酶纯酶制备:采用ge公司的histrap镍柱(histraptmhp,5ml)对步骤(3)得到的粗酶液进行分离纯化,并用pall公司的10k的超滤离心管进行超滤除盐。所述的纯化过程采用的纯化柱为能够特异性纯化带有his-tagged标签的蛋白质的histraphp柱,其步骤包括平衡、上样、平衡、洗脱、柱子再生;收集洗脱的部分并利用超滤离心管进行除盐;除盐后获得的液体即为纯化的带his-tag标签的胺脱氢酶的纯酶溶液。
(6)酶催化:胺脱氢酶的活性测定反应体系包含40mmnadh,0.2mol/lg-氢氧化钠缓冲溶液(ph8)、0.2mol/lnh4cl,50mm的5-甲基-2-己酮和20mg/ml酶液,在反应温度60℃下,250rpm震荡反应,反应时间30h。分离与检测配合5-甲基-2-己胺标准样品进行,其余具体步骤同实施例1步骤(7),结果如图2所示,利用酶催化不对称还原胺化得到产物r-5-甲基-2-己胺,产物胺收率为91.7%,e.e.值为93.2%
实施例3
构建单点突变(n262v)的氨基酸脱氢酶突变体,对262位氨基酸残基进行突变,具体步骤如下:
(1)引入突变:根据seqidno.02所示的核苷酸设计引物,设计包含有不同位点的上游引物和下游引物,所述上游引物和下游引物如下:
上游引物(seqidno.07):n262v-fgtaggaagtgccgtcaatcagctc
下游引物(seqidno.08):n262v-rgacggcacttcctacgatcgcttt
将引物和模板质粒混合后,加入高保真taq聚合酶kod-plus,进行全质粒pcr扩增,pcr结束后电泳检测pcr产物。
(2)构建可表达带his-tag标签的苯丙氨酸脱氢酶的重组表达菌株:苯丙氨酸脱氢酶(phedh)基因的原始序列来自bacillusnanhaiensis,如seqidno.02所示,原始氨基酸序列如seqidno.01所示;根据上述原始序列,设计如seqidno.07和seqidno.08所示的引物,采用pcr法构建5’端和3’端分别带有ndei和xhol酶切位点的苯丙氨酸脱氢酶基因,pcr合成过程由上海生工生物工程技术服务有限公司完成。pcr扩增产物经1%琼脂糖凝胶电泳鉴定后,胶回收phedh基因片段,用ndei和xhoi酶切酶进行双酶切,回收酶切产物,与同样双酶切的pet-28a质粒(带有his-tag标签)进行连接,连接好的质粒转化到大肠杆菌bl21(de3),得到pet28a-phedh质粒。将上述质粒转化至e.colibl21(de3),得到可表达带his-tag标签的胺脱氢酶的重组表达菌株e.colibl21(de3)/pet28a。
(3)重组大肠杆菌e.colibl21(de3)/pet28a的培养:以1%的接种量,将菌种接入200mllb培养基中。lb培养基的组成为10.0g/l胰蛋白胨,5.0g/l酵母浸膏,10g/lnacl。培养条件为:起始ph7.0,装液量体积分数为10%,培养温度37℃,摇床转速200rpm,培养时间6h。加入诱导剂iptg,使其终浓度为10mg/ml,继续在25℃、200rpm条件下培养12h。
(4)全细胞催化:以步骤3)得到的25mg/l的细胞液为基础配制全细胞催化体系:0.01mol/l苯乙酮,0.02mol/l氨水,0.01mol/l异丙醇,0.2mol/l的tris-盐酸缓冲液,25mg/l的细胞,反应体系ph7.5,反应体积5l,在反应温度10℃下,300rpm震荡反应,反应时间180h,利用全细胞催化不对称还原胺化得到产物r-苯乙胺。分离与检测如实施例1步骤7所示,测定产物胺收率为60.1%,e.e.值为95.7%。
实施例4
构建单点突变(y285l)的氨基酸脱氢酶突变体,对285位氨基酸残基进行突变,具体步骤如下:
(1)引入突变:根据seqidno.02所示的核苷酸设计引物,设计包含有不同位点的上游引物和下游引物,所述上游引物和下游引物如下:
上游引物(seqidno.09):y285l-ftacggacccgatctcatcgtgaac
下游引物(seqidno.10):y285l-rgagatcgggtccgtataaaattcc
将引物和模板质粒混合后,加入高保真taq聚合酶kod-plus,进行全质粒pcr扩增,pcr结束后电泳检测pcr产物。
(2)构建可表达带his-tag标签的苯丙氨酸脱氢酶的重组表达菌株:苯丙氨酸脱氢酶(phedh)基因的原始序列来自bacillusnanhaiensis,如seqidno.02所示,原始氨基酸序列如seqidno.01所示;根据上述原始序列,设计如seqidno.09和seqidno.10所示的引物,采用pcr法构建5’端和3’端分别带有ndei和xhol酶切位点的苯丙氨酸脱氢酶基因,pcr合成过程由上海生工生物工程技术服务有限公司完成。pcr扩增产物经1%琼脂糖凝胶电泳鉴定后,胶回收phedh基因片段,用ndei和xhoi酶切酶进行双酶切,回收酶切产物,与同样双酶切的pet-28a质粒(带有his-tag标签)进行连接,连接好的质粒转化到大肠杆菌bl21(de3),得到pet28a-phedh质粒。将上述质粒转化至e.colibl21(de3),得到可表达带his-tag标签的胺脱氢酶的重组表达菌株e.colibl21(de3)/pet28a。
(3)重组大肠杆菌e.colibl21(de3)/pet28a的培养:以1%的接种量,将菌种接入200mllb培养基中。lb培养基的组成为10.0g/l胰蛋白胨,5.0g/l酵母浸膏,10g/lnacl。培养条件为:起始ph7.0,装液量体积分数为10%,培养温度37℃,摇床转速200rpm,培养时间6h。加入诱导剂iptg,使其终浓度为10mg/ml,继续在25℃、200rpm条件下培养12h。
(4)粗酶液的制备:培养结束获得的发酵液,在冷冻离心机中离心(4℃,8000rpm,15min)获得细胞,弃上清液,沉淀用磷酸缓冲液(ph=7)重悬,充分洗涤后离心,重复操作3次,用磷酸缓冲液(ph7)配制成浓度为50g/l的细胞悬液。将制备的细胞悬液置于冰浴中,利用超声破碎仪对细胞液进行处理,细胞破碎仪探头置于液面下1cm,破碎条件为超声3秒,间隔6秒,超声60次,功率200w。然后4℃、12,000rpm离心15min去除不溶性细胞碎片,上清即为含有带his-tag标签的胺脱氢酶的粗酶液。
(5)胺脱氢酶纯酶制备:采用ge公司的histrap镍柱(histraptmhp,5ml)对步骤(3)得到的粗酶液进行分离纯化,并用pall公司的10k的超滤离心管进行超滤除盐。所述的纯化过程采用的纯化柱为能够特异性纯化带有his-tagged标签的蛋白质的histraphp柱,其步骤包括平衡、上样、平衡、洗脱、柱子再生;收集洗脱的部分并利用超滤离心管进行除盐;除盐后获得的液体即为纯化的带his-tag标签的胺脱氢酶的纯酶溶液。
(6)酶活力的检测方法:胺脱氢酶的活性测定反应体系包含10μl,40mmnadh、160μl,0.2mol/lg-氢氧化钠缓冲溶液(ph9.5)、10μl,40mm的2-丁酮和20mg/ml酶液,45℃下,200rpm震荡反应,反应时间30h,催化不对称还原胺化得到产物手性胺r-2-丁胺。分离与检测如实施例1步骤7所示,测定产物胺的收率为75.2%,e.e.值为97.7%。
实施例5
构建单点突变(n262l)的氨基酸脱氢酶突变体,对262位氨基酸残基进行突变,具体步骤如下:
上游引物(seqidno.11):n262l-fgtaggaagtgccctcaatcagctc
下游引物(seqidno.12):n262l-rgagggcacttcctacgatcgcttt
(2)构建可表达带his-tag标签的苯丙氨酸脱氢酶的重组表达菌株:苯丙氨酸脱氢酶(phedh)基因的原始序列来自bacillusnanhaiensis,如seqidno.02所示,原始氨基酸序列如seqidno.01所示;根据上述原始序列,设计如seqidno.11和seqidno.12所示的引物,采用pcr法构建5’端和3’端分别带有ndei和xhol酶切位点的苯丙氨酸脱氢酶基因,pcr合成过程由上海生工生物工程技术服务有限公司完成。pcr扩增产物经1%琼脂糖凝胶电泳鉴定后,胶回收phedh基因片段,用ndei和xhoi酶切酶进行双酶切,回收酶切产物,与同样双酶切的pet-28a质粒(带有his-tag标签)进行连接,连接好的质粒转化到大肠杆菌bl21(de3),得到pet28a-phedh质粒。将上述质粒转化至e.colibl21(de3),得到可表达带his-tag标签的胺脱氢酶的重组表达菌株e.colibl21(de3)/pet28a。
(3)重组大肠杆菌e.colibl21(de3)/pet28a的培养:以1%的接种量,将菌种接入200mllb培养基中。lb培养基的组成为10.0g/l胰蛋白胨,5.0g/l酵母浸膏,10g/lnacl。培养条件为:起始ph7.0,装液量体积分数为10%,培养温度37℃,摇床转速200rpm,培养时间6h。加入诱导剂iptg,使其终浓度为10mg/ml,继续在25℃、200rpm条件下培养12h。
(4)粗酶液的制备:培养结束获得的发酵液,在冷冻离心机中离心(4℃,8000rpm,15min)获得细胞,弃上清液,沉淀用磷酸缓冲液(ph=7)重悬,充分洗涤后离心,重复操作3次,用磷酸缓冲液(ph7)配制成浓度为50g/l的细胞悬液。将制备的细胞悬液置于冰浴中,利用超声破碎仪对细胞液进行处理,细胞破碎仪探头置于液面下1cm,破碎条件为超声3秒,间隔6秒,超声60次,功率200w。然后4℃、12,000rpm离心15min去除不溶性细胞碎片,上清即为含有带his-tag标签的胺脱氢酶的粗酶液。
(5)胺脱氢酶纯酶制备:采用ge公司的histrap镍柱(histraptmhp,5ml)对步骤(3)得到的粗酶液进行分离纯化,并用pall公司的10k的超滤离心管进行超滤除盐。所述的纯化过程采用的纯化柱为能够特异性纯化带有his-tagged标签的蛋白质的histraphp柱,其步骤包括平衡、上样、平衡、洗脱、柱子再生;收集洗脱的部分并利用超滤离心管进行除盐;除盐后获得的液体即为纯化的带his-tag标签的胺脱氢酶的纯酶溶液。
(6)酶催化:0.04mol/l2-羟基-3-丁酮,0.4mol/l丙氨酸,0.05mol/lnadh,0.2mol/l的碳酸钠缓冲液,酶100mg/ml,反应体积10ml,反应体系ph9.0,在反应温度50℃下,300rpm震荡反应,反应时间30h,催化不对称还原胺化得到产物r-2-羟基-3-丁胺。分离与检测如实施例1步骤7所示,测定产物胺收率为80.1%,e.e.值为96.7%。
实施例6
构建单点突变(y285m)的氨基酸脱氢酶突变体,对285位氨基酸残基进行突变,具体步骤如下:
(1)引入突变:根据seqidno.02所示的核苷酸设计引物,设计包含有不同位点的上游引物和下游引物,所述上游引物和下游引物如下:
上游引物(seqidno.13):y285m-ftacggacccgatatgatcgtgaac
下游引物(seqidno.14):y285m-rcatatcgggtccgtataaaattcc
将引物和模板质粒混合后,加入高保真taq聚合酶kod-plus,进行全质粒pcr扩增,pcr结束后电泳检测pcr产物。
(2)构建可表达带his-tag标签的苯丙氨酸脱氢酶的重组表达菌株:苯丙氨酸脱氢酶(phedh)基因的原始序列来自bacillusnanhaiensis,如seqidno.02所示,原始氨基酸序列如seqidno.01所示;根据上述原始序列,设计如seqidno.13和seqidno.14所示的引物,采用pcr法构建5’端和3’端分别带有ndei和xhol酶切位点的苯丙氨酸脱氢酶基因,pcr合成过程由上海生工生物工程技术服务有限公司完成。pcr扩增产物经1%琼脂糖凝胶电泳鉴定后,胶回收phcdh基因片段,用ndei和xhoi酶切酶进行双酶切,回收酶切产物,与同样双酶切的pet-28a质粒(带有his-tag标签)进行连接,连接好的质粒转化到大肠杆菌bl21(de3),得到pet28a-phedh质粒。将上述质粒转化至e.colibl21(de3),得到可表达带his-tag标签的胺脱氢酶的重组表达菌株e.colibl21(de3)/pet28a。
(3)重组大肠杆菌e.colibl21(de3)/pet28a的培养:以1%的接种量,将菌种接入200mllb培养基中。lb培养基的组成为10.0g/l胰蛋白胨,5.0g/l酵母浸膏,10g/lnacl。培养条件为:起始ph7.0,装液量体积分数为10%,培养温度37℃,摇床转速200rpm,培养时间6h。加入诱导剂iptg,使其终浓度为10mg/ml,继续在25℃、200rpm条件下培养12h。
(4)粗酶液的制备:培养结束获得的发酵液,在冷冻离心机中离心(4℃,8000rpm,15min)获得细胞,弃上清液,沉淀用磷酸缓冲液(ph=7)重悬,充分洗涤后离心,重复操作3次,用磷酸缓冲液(ph7)配制成浓度为50g/l的细胞悬液。将制备的细胞悬液置于冰浴中,利用超声破碎仪对细胞液进行处理,细胞破碎仪探头置于液面下1cm,破碎条件为超声3秒,间隔6秒,超声60次,功率200w。然后4℃、12,000rpm离心15min去除不溶性细胞碎片,上清即为含有带his-tag标签的胺脱氢酶的粗酶液。
(5)胺脱氢酶纯酶制备:采用ge公司的histrap镍柱(histraptmhp,5ml)对步骤(3)得到的粗酶液进行分离纯化,并用pall公司的10k的超滤离心管进行超滤除盐。所述的纯化过程采用的纯化柱为能够特异性纯化带有his-tagged标签的蛋白质的histraphp柱,其步骤包括平衡、上样、平衡、洗脱、柱子再生;收集洗脱的部分并利用超滤离心管进行除盐;除盐后获得的液体即为纯化的带his-tag标签的胺脱氢酶的纯酶溶液。
(6)酶催化:以步骤2)得到的300mg/l的酶为基础配制酶催化体系:0.04mol/l3-羟基-5-庚酮,0.2mol/lnh4no3,0.001-0.2mol/l甲酸,0.01mol/l的tris-盐酸缓冲液,300mg/l的酶,反应体系ph11,在反应温度10℃下,100-300rpm震荡反应,反应时间90h。分离与检测如实施例1步骤7所示,测定酶催化不对称还原胺化得到产物r-3-羟基-5-庚胺。产物胺收率为89.2%,e.e.值为90.5%。
实施例7
在n262v构建叠加突变(m333d)的氨基酸脱氢酶突变体,对262,333位氨基酸残基进行突变,具体步骤如下:
(1)引入突变:根据seqidno.02所示的核苷酸设计引物,设计包含有不同位点的上游引物和下游引物,所述上游引物和下游引物如下:
上游引物(seqidno.15):m333d-fcagatatctacggatgaagcagca
下游引物(seqidno.16):m333d-ratccgtagatatctggttctttgt
将引物和模板质粒混合后,加入高保真taq聚合酶kod-plus,进行全质粒pcr扩增,pcr结束后电泳检测pcr产物。
(2)构建可表达带his-tag标签的苯丙氨酸脱氢酶的重组表达菌株:苯丙氨酸脱氢酶(phedh)基因的原始序列来自bacillusnanhaiensis,如seqidno.02所示,原始氨基酸序列如seqidno.01所示;根据上述原始序列,设计如seqidno.15和seqidno.16所示的引物,采用pcr法构建5’端和3’端分别带有ndei和xhol酶切位点的苯丙氨酸脱氢酶基因,pcr合成过程由上海生工生物工程技术服务有限公司完成。pcr扩增产物经1%琼脂糖凝胶电泳鉴定后,胶回收phedh基因片段,用ndei和xhoi酶切酶进行双酶切,回收酶切产物,与同样双酶切的pet-28a质粒(带有his-tag标签)进行连接,连接好的质粒转化到大肠杆菌bl21(de3),得到pet28a-phedh质粒。将上述质粒转化至e.colibl21(de3),得到可表达带his-tag标签的胺脱氢酶的重组表达菌株e.colibl21(de3)/pet28a。
(3)重组大肠杆菌e.colibl21(de3)/pet28a的培养:以1%的接种量,将菌种接入200mllb培养基中。lb培养基的组成为10.0g/l胰蛋白胨,5.0g/l酵母浸膏,10g/lnacl。培养条件为:起始ph7.0,装液量体积分数为10%,培养温度37℃,摇床转速200rpm,培养时间6h。加入诱导剂iptg,使其终浓度为10mg/ml,继续在25℃、200rpm条件下培养12h。
(4)全细胞催化:以步骤3)得到的细胞液为基础配制全细胞催化体系:0.1mol/l3-己酮,0.2mol/l丙氨酸,0.2mol/l甲酸,0.2mol/l的tris-盐酸缓冲液,1000mg/l的细胞,反应体系ph7.5,反应体积5l,在反应温度25℃下,300rpm震荡反应,反应时间180h,利用全细胞催化不对称还原胺化得到产物r-3-己胺。分离与检测如实施例1步骤7所示,产物胺收率为78.1%,e.e.值为96.4%。
以上所述,仅为本发明的较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。
序列表
<110>厦门大学
<120>一种胺脱氢酶及其应用
<160>16
<170>siposequencelisting1.0
<210>1
<211>366
<212>prt
<213>bacillusnanhaiensis
<400>1
metpheglulysileserglnhisgluglnvalvalphecysasnasp
151015
proserthrglyleulysalaileilealailehisasnthrthrleu
202530
glyproalaleuglyglycysargmetargprotyrglyservalasp
354045
glualaleugluaspvalleuargleuserlysglymetthrtyrlys
505560
cysalaglyalaaspvalasppheglyglyglylysservalileile
65707580
glyaspprometthraspargthrprogluleupheargalaphegly
859095
glnphevalaspserleuasnglyargphetyrthrglythraspmet
100105110
glythrthrproaspaspphemethisalaleulysgluthrasncys
115120125
ilevalglyvalprogluglutyrglyglyserglyaspserserval
130135140
prothralaglnglyvaliletyrglyleuglnalathrileglnthr
145150155160
leugluglythraspgluleuserglylyssertyrserileglngly
165170175
leuglylysvalglyphelysvalalagluglnleuleualaalagly
180185190
thrglniletyrvalthraspileasnglulysalaleulysmetile
195200205
glngluargalagluleuleuproglyasnvalgluvalvalglugly
210215220
seraspiletyrglyvalaspalaaspilepheileprocysalaleu
225230235240
glyglyileilehisaspgluthrilegluglnleulysvallysala
245250255
ilevalglyseralaasnasnglnleuleugluasplyshisglyleu
260265270
tyrleuglnglnlysglyileleutyrglyproasptyrilevalasn
275280285
alaglyglyleuileglnvalalaaspgluleutyrglyproasnlys
290295300
alaargvalleuthrlysthrargalailetyraspserleuilegln
305310315320
iletyrsergluserthrlysasnglnileserthrmetglualaala
325330335
asnleuphecysgluglulysleuleualaargserlysargasnser
340345350
phephealahisasnargargprolystrpglnvalarghis
355360365
<210>2
<211>1076
<212>dna
<213>bacillusnanhaiensis
<400>2
agcaagttgtgttttgtaacgatccatcaacaggtctcaaggcaattatcgctatacata60
acacaacattaggcccagcactcggcggatgcagaatgagaccgtatggatcggttgatg120
aggcacttgaggatgtgctgagattgtcaaaaggtatgacatacaaatgcgctggtgcag180
atgttgacttcggtggaggtaaatcggtcatcatcggggatccgatgacggatcgtacac240
cagagttgttccgagcattcggacagtttgtagattcattaaacggtcgcttttatacag300
gaacagatatgggaacaacacctgatgattttatgcacgcgttaaaagaaacgaattgta360
tcgtcggggtacccgaagaatatggcggcagcggcgattcttctgttccaacagcacaag420
gagttatatacggacttcaagctaccattcagacgcttgaaggaacagatgaactttcag480
gtaagtcatactctatacaaggtttaggaaaagtaggttttaaggtcgcagagcaactgc540
tcgcagccggtacacaaatctatgttactgatattaatgaaaaagcattaaagatgattc600
aagaacgagcagaactcctacctggaaatgtggaagtagttgaaggaagcgacatctacg660
gggtggatgctgatattttcattccttgcgcactcggcggaatcattcacgatgaaacaa720
ttgaacaactaaaagtaaaagcgatcgtaggaagtgccaacaatcagctcttagaagata780
agcacggactttatttgcagcaaaaaggaattttatacggacccgattatatcgtgaacg840
caggagggcttattcaggtagctgatgagctttatggaccgaataaagcccgtgtattaa900
cgaaaacgagagcgatctatgacagtctgatacagatttatagcgagagtacaaagaacc960
agatatctacgatggaagcagcaaatcttttctgtgaggagaagcttttggcccgctcaa1020
aacgaaacagctttttcgctcacaaccgaagaccaaaatggcaggttagacattaa1076
<210>3
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>3
aattgtatcgtcctcgtacccgaa24
<210>4
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>4
gaggacgatacaattcgtttcttt24
<210>5
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>5
aattgtatcgtcatggtacccgaa24
<210>6
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>6
catgacgatacaattcgtttcttt24
<210>7
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>7
gtaggaagtgccgtcaatcagctc24
<210>8
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>8
gacggcacttcctacgatcgcttt24
<210>9
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>9
tacggacccgatctcatcgtgaac24
<210>10
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>10
gagatcgggtccgtataaaattcc24
<210>11
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>11
gtaggaagtgccctcaatcagctc24
<210>12
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>12
gagggcacttcctacgatcgcttt24
<210>13
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>13
tacggacccgatatgatcgtgaac24
<210>14
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>14
catatcgggtccgtataaaattcc24
<210>15
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>15
cagatatctacggatgaagcagca24
<210>16
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>16
atccgtagatatctggttctttgt24
- 还没有人留言评论。精彩留言会获得点赞!