复制蛋白A靶向的铂类化合物的制作方法
复制蛋白a靶向的铂类化合物
技术领域
[0001]
本发明属于铂类化合物技术领域,更加具体地说,涉及一类基于dna损伤修复的铂类化合物抗肿瘤活性、减轻毒副作用及克服临床铂类药物耐药的研究。
背景技术:
[0002]
恶性肿瘤,时刻威胁着人类健康及生命安全,对恶性肿瘤的治疗研究具有十分重要的价值和意义。提高化疗药物的靶向性、选择性及安全性,是当前新型抗肿瘤药物的研发趋势。以顺铂(cisplatin,cddp)为代表的铂类药物在临床肿瘤治疗(如卵巢癌、非小细胞肺癌)中起到了至关重要的作用,50%以上的肿瘤化疗需要铂类药物的参与。但是,肿瘤耐药极大地限制了该类药物的发展。80%以上的上皮性卵巢癌患者由于铂药耐药从而导致肿瘤复发,化疗失败。
[0003]
从铂类药物作用的分子机制我们可知,由于铂类药物的作用靶点为dna,可与dna发生链间或链内交联,形成dna加合物,致使dna损伤,阻碍dna复制、转录,最终引起细胞凋亡。因此,dna损伤修复与铂类药物的化疗效果、肿瘤耐药密切相关。研究表明,核苷切除修复(nucleotide excision repair,ner)与同源重组修复(homologous recombination,hr)可大大降低铂类药物的化疗效果,因此,选择性地对二者进行抑制,可进一步增强肿瘤细胞对铂类药物的化疗敏感性。
[0004]
复制蛋白a(replication protein a,rpa)是真核细胞中主要的单链结合蛋白,包含rpa1,rpa2和rpa3三个亚单位,在dna复制和损伤修复过程中起着重要的作用。dna复制时,rpa具有解链、结合单链模板并维持dna连续复制的功能;dna损伤时,rpa作为重要的dna损伤修复蛋白,在核苷酸切除修复(ner)、dna错配修复(mmr)、同源重组修复(hr)等修复过程发挥重要作用。同时,rpa与具有染色体结构维持、保护、修复功能的蛋白质聚集在dna损伤位点,共同完成对dna损伤的检测并进行修复。同时,dna损伤同源修复蛋白rad51,rad52与rpa-ssdna协同作用,共同完成dna的损伤修复。rpa对ner和hr等修复方式的作用,使得铂类化疗药物的疗效的降低,肿瘤细胞的耐药性增强(shuck s c,turchi j j.targeted inhibition of rpa reveals cytotoxic activity,synergy with chemotherapeutic dna damaging agents and insight into cellular function[j].cancer research,2010,70(8):3189.)。近年来rpa抑制剂的发现让恶性肿瘤患者重新看到了曙光。而rpa抑制剂和铂类药物的结合将对提高抗肿瘤活性及抗肿瘤耐药具有十分重要的意义。
技术实现要素:
[0005]
本发明的目的在于克服现有铂类药物不足,提供新型铂类(iv)rpa抑制剂前药的制备方法与应用,与临床药物顺铂相比,本发明铂类抗肿瘤化合物(即新型铂类(iv)rpa抑制剂前药)表现出显著优于顺铂的体内、体外抗肿瘤活性,并具有降低系统毒性、克服顺铂耐药的优点。
[0006]
将rpa小分子抑制剂与铂药结合形成新型铂类(iv)rpa小分子抑制剂前药具有以
下优势:(1)从化学结构角度来看,新型铂类化合物改变了先前化合物的极性、log p、跨膜转运速率等物理化学性质,进而改变铂类药物的摄取方式,以增加细胞内铂含量;同时,用pt(iv)八面体化学构型的化学惰性,从而降低铂类药物的反应活性及毒性,并在细胞内还原、释放协同作用分子,提高了铂类化疗疗效。(2)从作用靶点角度来看,rpa抑制剂阻断了rpa-dna相互作用。在dna复制中,rpa抑制剂利用癌细胞的高度增殖性质,使细胞阻滞在s期,进而造成细胞凋亡。因此,利用复制蛋白a小分子抑制剂的靶向性、协同性特点,从而降低铂药被dna修复机制的修复,克服了肿瘤细胞对铂类药物的耐药性。
[0007]
本发明的技术目的通过下述技术方案予以实现:
[0008]
基于rpa抑制剂的铂类化合物(即复制蛋白a靶向的铂类化合物),具有如下化学式所示结构:
[0009][0010]
其中r1为i或br;r2为氢原子、丁酰基、己酰基、辛酰基、十一碳酰基、十六碳酰基;n为2或3。优选r1为i;r2为氢原子或者己酰基;n为2或者3。
[0011][0012]
就本申请而言,以四价顺铂前药为母体,轴向位置先后结合rpa小分子抑制剂和脂肪链来合成目标化合物。
efficacy of pt-based chemotherapy in lung and ovarian cancer[j].biochemical pharmacology,2015,93(1):25-33.)。
[0019]
上述铂类化合物在制备抗肿瘤药物中的应用。与现有技术相比,本发明铂化合物在hela、a549、nci-h460等细胞系中抗肿瘤活性明显优于顺铂等经典铂类药物。本发明铂化合物在nci-h460/cis耐顺铂细胞系中仍然保持优良的抗肿瘤活性。本发明新型铂类(iv)rpa小分子抑制剂前药可在造成dna损伤的同时抑制ner(核苷酸切除修复)和hrr(同源重组修复)两种修复方式对dna的修复。在体内抗肿瘤实验中,本发明具有良好的抗肿瘤活性和抗肿瘤耐药特性,同时降低了铂类药物的毒性。
附图说明
[0020]
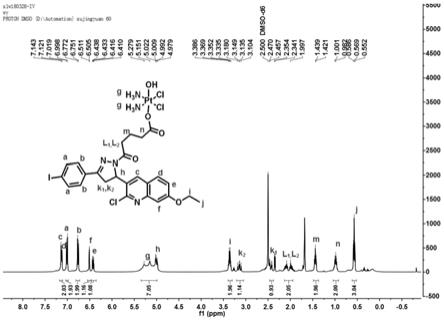
图1是本发明实施例中制备的物质3核磁共振氢谱图。
[0021]
图2是本发明实施例中制备的物质3核磁共振碳谱图。
[0022]
图3是本发明实施例中制备的物质3液相纯度检测图谱。
[0023]
图4是本发明实施例中制备的物质3高分辨质谱图。
[0024]
图5是本发明实施例中制备的物质3a核磁共振氢谱图。
[0025]
图6是本发明实施例中制备的物质3a核磁共振碳谱图。
[0026]
图7是本发明实施例中制备的物质3a液相纯度检测图谱。
[0027]
图8是本发明实施例中制备的物质3b核磁共振氢谱图。
[0028]
图9是本发明实施例中制备的物质3b核磁共振碳谱图。
[0029]
图10是本发明实施例中制备的物质3b高分辨质谱图。
[0030]
图11是本发明实施例中制备的物质3b液相纯度检测图谱。
[0031]
图12是本发明实施例中制备的物质3c核磁共振氢谱图。
[0032]
图13是本发明实施例中制备的物质3c核磁共振碳谱图。
[0033]
图14是本发明实施例中制备的物质3c液相纯度检测图谱。
[0034]
图15是本发明实施例中制备的物质3c高分辨质谱图。
[0035]
图16是本发明实施例中制备的物质3d核磁共振氢谱图。
[0036]
图17是本发明实施例中制备的物质3d核磁共振碳谱图。
[0037]
图18是本发明实施例中制备的物质3d液相纯度检测图谱。
[0038]
图19是本发明实施例中制备的物质3e核磁共振氢谱图。
[0039]
图20是本发明实施例中制备的物质3e核磁共振碳谱图。
[0040]
图21是本发明实施例中制备的物质3e液相纯度检测图谱。
[0041]
图22是本发明实施例中制备的物质4核磁共振氢谱图。
[0042]
图23是本发明实施例中制备的物质4核磁共振碳谱图。
[0043]
图24是本发明实施例中制备的物质4高分辨质谱图。
[0044]
图25是本发明实施例中制备的物质5核磁共振氢谱图。
[0045]
图26是本发明实施例中制备的物质5核磁共振碳谱图。
[0046]
图27是本发明实施例中制备的物质5高分辨质谱图。
[0047]
图28是本发明实施例中制备的物质6核磁共振氢谱图。
[0048]
图29是本发明实施例中制备的物质6核磁共振碳谱图。
[0049]
图30是本发明实施例中制备的物质6高分辨质谱图。
[0050]
图31是本发明实施例中制备的3b对细胞周期、凋亡结果示意图。
[0051]
图32是本发明实施例中制备的3b对体内抗肿瘤实验结果示意图。
具体实施方式
[0052]
下面结合具体实施例进一步说明本发明的技术方案。实验仪器如下:核磁共振测定仪,瑞士bruker公司avance iii 400mhz;高分辨质谱,美国安捷伦公司agilent 6520q-tof lc/ms;高效液相色谱仪,日本岛津公司spd-20a;激光共聚焦显微镜,日本奥林巴斯fv-1000;电子分析天平,德国sartorius bp211d;旋转蒸发仪,上海申生科技有限公司r204;恒温磁力搅拌器,河南巩义予华仪器有限公司85-2。原料和试剂如下:顺铂,山东铂源药业有限公司,化学纯;tbtu,天津希恩斯生化科技有限公司,化学纯;30%h2o2,天津北联化学有限公司,化学纯;超干n,n
’-
二甲基甲酰胺和超干n,n
’-
二甲基亚砜,百灵威科技有限公司,化学纯;二氯甲烷,天津市津东天正精细化学试剂厂,分析纯;无水乙醚,天津市康科德科技有限公司,分析纯;甲醇,天津市康科德科技有限公司,色谱纯;人非小细胞肺癌细胞系a549,人非小细胞肺癌细胞系nci-h460和人宫颈癌细胞系hela,美国atcc;人非小细胞肺癌细胞系nci-h460cisr,北京协和细胞资源中心;dmem培养基和rpmi 1640培养基,美国gibco公司;胎牛血清,美国hyclone公司。
[0053]
实施例1—基于新型铂类pt(iv)rpa小分子抑制剂前药的合成及表征,如附图1—30所示
[0054]
精确称取顺铂(100mg,0.34mmol)于10ml圆底烧瓶中,缓慢逐滴加入2ml 30%h2o2,反应液于75℃避光回流5h后,将反应液避光静置于4℃条件过夜。静置后,将混悬液采用离心分离得黄色固体沉淀,沉淀经蒸馏水、乙醇和乙醚洗涤后,真空干燥,得到物质2黄色固体粉末89mg,产率88.28%。
[0055][0056]
精确称取物质2(50.11mg,0.15mmol)于25ml圆底烧瓶中,加入dmso溶解,反应液于60℃氩气保护下避光搅拌30min至溶液澄清。另精确称取物质tdrl-551(0.15mmol)和tbtu(0.30mmol),加dmso溶解,向反应液中加入70μl三乙胺(0.6mmol),室温下超声反应30min,将反应液加入至物质2圆底烧瓶中反应。反应液于60℃氩气保护下避光搅拌12h,得红褐色澄清液体。将反应液水洗,离心分离得淡黄色固体沉淀。沉淀以二氯甲烷-甲醇(15:1)为展开剂,薄层色谱纯化,得到物质3淡黄色固体粉末。产量:45.5mg,产率:33.4%,纯度:
97.16%。1h nmr(400mhz,dmso-d6,ppm),δ7.96(d,j=8.9hz,1h),7.83(d,j=8.4hz,1h),7.59(d,j=8.4hz,1h),7.33(d,j=2.1hz,1h),7.25(dd,j=9.0,2.3hz,1h),6.29-5.75(m,4h),4.19(q,j=6.9hz,1h),3.97(dd,j=18.0,12.1hz,1h),3.21(dd,j=28.3,5.2hz,1h),2.86(dtd,j=30.5,15.4,7.5hz,1h),2.26(t,j=7.3hz,1h),1.91-1.71(m,1h),1.39(t,j=6.9hz,1h);
13
c nmr(101mhz,dmso-d6,ppm):δppm 181.21,170.98,160.97,154.22,148.77,138.18,131.13,130.58,129.97,129.24,122.66,120.86,107.24,97.89,64.30,41.06,36.46,33.58,21.63,15.02.hr-ms(esi,positive-ion mode),m/z:calcd for c
25
h
29
cl3in5o5pt[m+h]
+
:905.9927;found:905.9911.
[0057][0058]
精确称取物质3(30mg,0.033mmol)置于25ml圆底烧瓶中,加入dmf溶解至澄清,向溶液中加入丁酸酐(10.80μl,0.066mmol),在氩气保护下,45℃避光反应24h,得淡黄色溶液。反应结束,后旋蒸除去dmf,薄层色谱纯化,得到物质3a淡黄色固体粉末。产量:20.10mg,产率:59.11%,纯度:96.14%。1h nmr(400mhz,dmso-d6,ppm)δ7.95(d,j=9.0hz,1h),7.84(d,j=8.3hz,1h),7.59(d,j=8.3hz,1h),7.34(d,j=1.9hz,1h),7.25(dd,j=9.0,2.2hz,1h),6.56(s,3h),5.83(dd,j=12.0,5.2hz,1h),4.18(q,j=6.8hz,1h),3.97(dd,j=18.1,12.2hz,1h),3.36-3.18(m,3h),3.03-2.66(m,2h),2.33(t,j=7.3hz,1h),2.20(t,j=7.3hz,1h),1.89-1.70(m,1h),1.47(dd,j=14.7,7.3hz,1h),1.39(t,j=6.9hz,2h),0.87(t,j=7.4hz,2h).
13
c nmr(101mhz,dmso-d6,ppm)δ180.76,180.36,170.20,160.33,153.68,148.13,137.54,130.45,129.94,129.31,128.60,122.02,120.25,106.57,97.38,63.66,62.05,40.41,37.61,35.00,32.74,20.73,18.83,14.39,13.66.
[0059][0060]
精确称取物质3(30mg,0.033mmol)置于25ml圆底烧瓶中,加入dmf溶解,搅拌至澄清,向溶液中加入己酸酐(15.20μl,0.066mmol),在氩气保护下,45℃避光搅拌24h,得淡黄色溶液。反应结束后,旋蒸除去dmf,经薄层色谱纯化,得物质3b淡黄色固体粉末,产量:23.30mg,产率:63.96%,纯度:98.68%。1h nmr(400mhz,dmso-d6,ppm)δ7.95(d,j=9.1hz,
1h),7.84(d,j=8.5hz,1h),7.59(d,j=8.5hz,1h),7.34(d,j=2.3hz,1h),7.25(dd,j=9.0,2.4hz,1h),6.88-6.33(m,3h),5.83(dd,j=12.0,5.3hz,1h),4.18(q,j=6.9hz,1h),4.04-3.85(m,1h),3.28(dd,j=18.1,5.2hz,1h),2.88(tdd,j=23.4,15.6,7.5hz,1h),2.33(t,j=7.4hz,1h),2.21(t,j=7.5hz,1h),1.90-1.70(m,1h),1.56-1.43(m,1h),1.39(d,j=7.0hz,1h),1.26(dd,j=7.1,3.9hz,2h),0.86(t,j=6.9hz,1h).
13
c nmr(101mhz,dmso-d6,ppm)δ180.87,180.37,170.32,170.20,160.23,153.60,148.02,137.54,130.63,129.93,129.21,128.55,121.91,120.55,106.92,97.59,63.66,35.94,35.65,32.85,31.12,24.80,22.00,20.65,14.39,13.74.hr-ms(esi)m/z calcd for c
31
h
40
cl3in5o6pt[m+h]
+
:1006.07156,found:1006.07227.
[0061][0062]
精确称取物质3(30mg,0.033mmol)置于25ml圆底烧瓶中,加入dmf溶解,搅拌至澄清,向溶液中加入辛酸酐(19.61μl,0.066mmol),在氩气保护下,45℃避光搅拌24h,溶液变为淡黄色溶液。反应结束后旋蒸除去反应液中dmf,薄层色谱纯化,得到物质3c淡黄色固体粉末,产量:20.5mg,产率:61.75%,纯度:97.96%。1h nmr(400mhz,dmso-d6,ppm)δ7.95(d,j=9.0hz,1h),7.84(d,j=8.3hz,1h),7.59(d,j=8.3hz,1h),7.34(s,1h),7.26(d,j=8.9hz,1h),6.56(s,2h),5.95-5.76(m,1h),4.35-4.07(m,1h),3.97(dd,j=18.0,12.1hz,1h),3.28(dd,j=18.2,5.1hz,1h),3.07-2.75(m,1h),2.33(t,j=7.1hz,1h),2.21(t,j=7.4hz,1h),1.89-1.70(m,1h),1.44(d,j=6.9hz,1h),1.39(t,j=6.9hz,1h),1.24(s,3h),0.86(t,j=6.5hz,1h).
13
c nmr(101mhz,dmso-d6,ppm)δ180.86,170.20,160.33,153.68,148.13,137.54,130.44,128.61,122.02,106.57,97.39,63.65,40.08,39.87,39.66,39.45,39.25,39.04,38.83,35.65,32.74,31.18,28.56,25.42,22.07,20.72,14.39,13.96.hr-ms(esi)m/z calcd for c
33
h
44
cl3in5o6pt[m+h]
+
:1034.10293,found:1034.10339.
[0063][0064]
精确称取物质3(30mg,0.033mmol)置于25ml圆底烧瓶中,加入dmf溶解,搅拌至澄清,向溶液中加入十二烷酸酐(25.25mg,0.066mmol),在氩气保护下,45℃避光搅拌24h,得
淡黄色溶液。反应结束后旋蒸除去反应液中dmf,薄层色谱纯化,得到物质3d淡黄色固体粉末,产量:20.5mg,产率:63.94%,纯度:96.54%。1h nmr(400mhz,dmso-d6,ppm)δ7.95(d,j=9.0hz,1h),7.84(d,j=8.4hz,1h),7.59(d,j=8.4hz,1h),7.34(d,j=2.1hz,1h),7.25(dd,j=9.0,2.3hz,1h),6.56(s,3h),5.83(dd,j=12.0,5.2hz,1h),4.18(q,j=6.8hz,1h),3.97(dd,j=18.0,12.1hz,1h),3.28(dd,j=18.1,5.1hz,1h),3.01-2.74(m,1h),2.33(t,j=7.3hz,1h),2.21(t,j=7.4hz,1h),1.94-1.74(m,1h),1.45(s,1h),1.40(dd,j=13.5,6.6hz,2h),1.23(s,8h),0.85(t,j=6.7hz,2h).
13
c nmr(101mhz,dmso-d6,ppm)δ180.87,180.36,170.19,160.33,153.66,148.13,137.54,130.44,129.92,129.30,128.59,122.01,120.24,106.56,97.37,63.65,35.67,35.01,32.75,31.28,29.04,29.02,28.97,28.92,28.71,28.61,25.43,22.08,20.73,14.39,13.95.
[0065][0066]
精确称取物质3(30mg,0.033mmol)置于25ml圆底烧瓶中,加入dmf溶解,搅拌至澄清,向溶液加入棕榈酸酐(32.65mg,0.066mmol),在氩气保护下,45℃避光搅拌24h,得淡黄色溶液。反应结束后旋蒸除去反应液中dmf,薄层色谱纯化,得到物质3e淡黄色固体粉末,产量:26.0mg,产率:68.73%,纯度:98.84%。1h nmr(400mhz,dmso-d6,ppm)δ7.96(d,j=8.8hz,1h),7.84(d,j=8.1hz,1h),7.59(d,j=8.2hz,1h),7.34(s,1h),7.26(d,j=9.0hz,1h),6.63(d,j=61.2hz,3h),5.83(dd,j=12.0,5.0hz,1h),4.19(d,j=6.9hz,1h),3.97(dd,j=17.7,12.1hz,1h),3.45(s,1h),3.28(dd,j=18.1,5.0hz,1h),3.06-2.72(m,1h),2.33(t,j=7.0hz,1h),2.21(t,j=7.3hz,1h),1.89-1.75(m,1h),1.45(s,1h),1.41(dd,j=16.5,9.7hz,2h),1.23(s,11h),0.85(t,j=6.3hz,1h).
13
c nmr(101mhz,dmso-d6,ppm)δ181.37,180.87,170.70,160.83,154.16,148.64,138.04,130.95,129.09,122.51,120.74,107.06,97.87,64.15,58.05,42.75,36.17,33.24,31.77,29.54,29.49,29.19,25.94,22.58,21.23,14.89,14.45.
[0067][0068]
精确称取物质2(50.11mg,0.15mmol)于25ml圆底烧瓶中,加入dmso溶解,反应液于氩气保护下60℃避光搅拌30min,溶液变为无色澄清。另取精确称取物质tdrl-543(0.15mmol)和tbtu(0.30mmol),加入dmso溶解,向反应液中加入70μl三乙胺,室温下超声震
荡反应30min,反应液加入物质2溶液中,将反应瓶于氩气保护下60℃避光搅拌12h,得红褐色澄清液体。将反应液水洗,离心分离得淡黄色固体沉淀。以二氯甲烷-甲醇(15:1)为展开剂,薄层色谱纯化,得到物质4淡黄色固体粉末。产量:38.3mg,产率:29.7%。1h nmr(400mhz,dmso-d6,ppm)δ7.96(d,j=8.5hz,1h),7.75(d,j=8.3hz,1h),7.66(d,j=8.4hz,1h),7.33(s,1h),7.25(d,j=8.9hz,1h),6.21-5.77(m,4h),4.19(dd,j=13.8,6.8hz,1h),3.98(dd,j=18.0,12.1hz,1h),3.22(dd,j=36.5,4.9hz,1h),2.98-2.73(m,1h),2.27(t,j=7.2hz,1h),1.82(dd,j=14.8,7.5hz,1h),1.39(t,j=6.9hz,2h);
13
c nmr(101mhz,dmso-d6,ppm)δ180.81,170.58,160.55,160.16,153.59,148.35,131.93,130.45,129.55,128.93,123.90,122.24,120.44,106.83,63.87,48.78,40.71,36.05,33.16,21.22,14.59;hr-ms(esi,positive-ion mode)m/z:calcd for c
25
h
29
brcl3n5o5pt[m+h]
+
:858.0065;found:858.0107.
[0069][0070]
精确称取物质2(50.11mg,0.15mmol)于25ml圆底烧瓶中,加入dmso溶解,反应液于氩气保护下60℃避光搅拌30min,溶液变为无色澄清。另取精确称取物质tdrl-505(0.15mmol)和tbtu(0.30mmol),加入dmso溶解,向反应液中加入70μl三乙胺,室温下超声震荡反应30min,反应液加入物质2溶液中,将反应瓶于氩气保护下60℃避光搅拌12h,得红褐色澄清液体。将反应液水洗,离心分离得淡黄色固体沉淀。以二氯甲烷-甲醇(13:1)为展开剂,薄层色谱纯化,得到物质5淡黄色固体粉末。产量:35.2mg,产率:27.7%。1h nmr(400mhz,dmso-d6,ppm)δ8.02(s,1h),7.91(d,j=8.9hz,1h),7.73(d,j=8.3hz,2h),7.66(d,j=8.2hz,2h),7.33(s,1h),7.25(d,j=8.9hz,1h),6.27-5.67(m,7h),4.18(d,j=6.9hz,2h),3.99(dd,j=17.8,12.2hz,1h),3.27(s,1h),3.03(d,j=44.8hz,2h),2.55(d,j=7.1hz,1h),1.39(t,j=6.8hz,3h);
13
c nmr(101mhz,dmso-d6,ppm)δ180.12,170.06,160.32,153.47,148.14,134.96,133.29,131.74,130.24,129.87,129.26,128.65,123.70,122.07,120.22,106.71,63.67,57.50,40.60,31.19,29.90,14.37;hr-ms(esi,positive-ion mode)m/z:calcd for c
24
h
27
brcl3n5o5pt[m+h]
+
:843.9099;found:843.9970.
[0071][0072]
精确称取物质2(50.11mg,0.15mmol)于25ml圆底烧瓶中,加入dmso溶解,反应液于氩气保护下60℃避光搅拌30min,溶液变为无色澄清。另取精确称取物质tdrl-550(0.15mmol)和tbtu(0.30mmol),加入dmso溶解,向反应液中加入70μl三乙胺,室温下超声震荡反应30min,反应液加入物质2溶液中,将反应瓶于氩气保护下60℃避光搅拌12h,得红褐
色澄清液体。将反应液水洗,离心分离得淡黄色固体沉淀。以二氯甲烷-甲醇(13:1)为展开剂,薄层色谱纯化,得到物质6淡黄色固体粉末。产量:34.8mg,产率:25.9%。1h nmr(400mhz,dmso-d6,ppm)δ8.01(s,1h),7.91(d,j=9.0hz,1h),7.83(d,j=8.3hz,2h),7.57(d,j=8.3hz,2h),7.33(s,1h),7.26(d,j=8.9,2.2hz,1h),6.32-5.70(m,7h),4.18(d,j=6.9hz,2h),3.98(dd,j=17.9,12.1hz,1h),3.27(dd,j=18.1,5.2hz,1h),3.15-2.87(m,2h),2.62-2.52(m,1h),1.39(t,j=6.9hz,3h);
13
c nmr(101mhz,dmso-d6,ppm)δ180.34,170.27,160.56,153.97,148.38,137.83,130.71,130.09,129.52,128.80,122.30,120.52,106.86,97.58,63.92,57.66,40.74,36.03,31.35,30.08,14.65;hr-ms(esi,positive-ion mode)m/z:calcd for c
24
h
27
cl3in5o5pt[m+h]
+
:891.9770;found:891.9813.
[0073]
实施例2—化合物(3a-3e)及顺铂的抗肿瘤活性评价
[0074]
mtt比色法是实验研究中用来检测细胞生长、存活的方法之一。在活细胞线粒体中外源性的mtt能够被细胞内的琥珀酸脱氢酶还原为一种紫蓝色且难溶于水的结晶物甲瓒,甲瓒沉积在细胞胞质中,但是死亡细胞不会出现此现象。当甲瓒遇到dmso时,沉积在细胞中的甲瓒会被dmso溶解。采用酶标仪检测570nm波长处的甲瓒光密度值(od值)来间接反映活细胞的数量,形成mtt结晶物的量与活细胞的数量成正相关,用graphpad prism 6软件处理数据计算ic
50
值并作图,ic
50
值指被测量药物对细胞生长的半抑制浓度。实验结果表明,如下表所示,本发明新型铂类(iv)rpa小分子抑制剂前药在hela、a549、nci-h460等细胞系中抗肿瘤活性明显优于顺铂等经典铂类药物。本发明新型铂类(iv)rpa小分子抑制剂前药在nci-h460cisr等耐顺铂细胞系中仍然保持优良的抗肿瘤活性。本发明系列前药和顺铂在不同细胞系中作用72h的ic
50
值
a
[0075][0076]
a
ic
50
值
±
标准差(μm)由倍半稀释方法,每组三个复孔,重复1次,给药孵育72h拟合量-效曲线得到。
b
fi(增长倍数)是ic
50
(cddp)与ic
50
(物质3b)的比值。
c
rf(耐药系数)ic
50
(nci-h460cisr)与ic
50
(nci-h460)的比值。
b
fi(增长倍数)是ic
50
(cddp)与ic
50
(物质3)的比值。
e
nd,未测定。
[0077]
实施例3
[0078]
我们利用细胞周期、凋亡实验来验证本发明新型铂类(iv)rpa小分子抑制剂前药的抗肿瘤活性。细胞周期是指细胞从一次有丝分裂完成开始到下一次有丝分裂结束为止的过程。它可分为三个时相:g0/g1期、s期、g2/m期。g0/g1期为有丝分裂的准备期,rna及相关蛋白质的合成加倍,为遗传物质dna的合成做必要的准备;s期为dna合成期,在此期完成所有dna的合成;g2/m期为蛋白质合成和有丝分裂期,将已合成好的染色体及其它成分平均分配到两个新形成的子细胞中,以上过程周而复始从而实现细胞的增殖。细胞周期在细胞增殖、凋亡的调控中具有十分重要意义。
[0079]
如附图31和32所示,细胞周期实验结果表明,本发明新型铂类(iv)rpa小分子抑制剂前药随着物质3b的浓度的递增(0.1-10μm),分布于g2和s期的细胞数量呈现上升趋势;同时,g1期细胞比例逐渐降低。这表明在药物作用后,细胞周期过程中,细胞聚集在g2期和s期,物质3b可使hela细胞在g2期和s期发生阻滞,处理组1μm浓度下物质3b对肿瘤细胞主要为s期阻滞,10μm浓度下转变为g2期阻滞。与对照组相比,3b对hela细胞的周期阻滞更为明显。细胞凋亡实验结果表明,与对照组顺铂,及单取代(3)和联合用药(551+pt)相比,处理组物质3b的凋亡率明显升高,而且随着给药浓度的升高,处理组的凋亡率显著增加,凋亡区域主要集中在晚凋区域。这说明,本发明新型铂类(iv)rpa小分子抑制剂前药3b能够有效的诱导hela细胞的凋亡。
[0080]
实施例4
[0081]
我们通过体内抗肿瘤实验进一步研究了本发明新型铂类(iv)rpa小分子抑制剂前药的体内抗肿瘤活性及抗耐药活性评价。以4龄的裸鼠为实验动物,以体外活性较为敏感的人肿瘤细胞株(hela)构建裸鼠皮下移植瘤模型,模型构建成功后,将荷瘤裸鼠随机分组,尾静脉注射给药,观察并记录动物体重及肿瘤体积,实验维持4周,实验结束后,采用颈椎脱臼法处死实验动物,并收集肿瘤组织及主要器官,用于he染色及作用机制分析。实验结果表明,在pbs,cddp,551+pt,tdrl-551,物质3和物质3b六组实验模型中,通过24天的实验周期,对比其他组别,物质3b处理组存在着明显的抑瘤效果,pbs,tdrl-551,551+pt和物质3四组裸鼠的肿瘤体积总体呈上升趋势。尽管cddp给药组也存在着明显的抑瘤效果,但cddp组裸鼠体重下降过于明显。说明cddp化合物对裸鼠本身产生较大的毒副作用。
[0082]
依照本发明内容进行工艺参数的调整,均可实现本发明铂类化合物的制备,且表现出与实施例基本一致的抗癌性能。以上对本发明做了示例性的描述,应该说明的是,在不脱离本发明的核心的情况下,任何简单的变形、修改或者其他本领域技术人员能够不花费创造性劳动的等同替换均落入本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1