有机化合物及包含其的有机发光二极管的制作方法
[0001]
本发明涉及有机功能材料技术领域,特别是涉及一种有机化合物及包含其的有机发光二极管。
背景技术:
[0002]
有机发光二极管(oleds)因其具有对比度高、广视角、能耗低、轻薄和可弯曲等优点已经成为高端显示的主流,比如高端旗舰手机、电视、照明、可穿戴显示等。未来oleds应用肯定会越来越广泛。虽然如此,oleds技术还有改善的空间,比如蓝光材料相比红绿发光材料,其效率和寿命还差很多。
[0003]
目前主要使用的蓝光材料为磷光材料,这类材料一般需要掺杂在主体材料中使用,这是因为它们具有严重的浓度猝灭效应。但是这种主客体材料搭配目前很少能同时得到高效率和高寿命的器件性能。另外蓝光材料具有高能隙,因此对主体材料的要求也很高,需要其具有比蓝光客体材料更高的能隙。目前高能隙的主体材料比较少,材料选择有限,且这种高能隙材料容易老化,从而降低oleds的使用寿命。
[0004]
传统的发光材料多为具有大π共轭体系的刚性平面分子,在稀溶液中有很高的荧光量子产率,但在固态下荧光减弱甚至发生猝灭,即聚集导致了荧光猝灭。造成这种现象的主要原因是:分子间的相互作用导致了非辐射能量转换或形成了不利于荧光发射的物种。而在实际应用中,荧光材料往往需要制成固体或薄膜形式,这也是传统发光材料需要掺杂在其它主体材料中使用的又一原因,由此增加了发光器件的制备成本和不稳定性因素。
技术实现要素:
[0005]
基于此,有必要提供一种有机化合物。该有机化合物不需要主体材料,也能够在固态条件下,实现高效率和高稳定性的蓝光发射,减少主客体材料掺杂的工艺不稳定性因素和成本。
[0006]
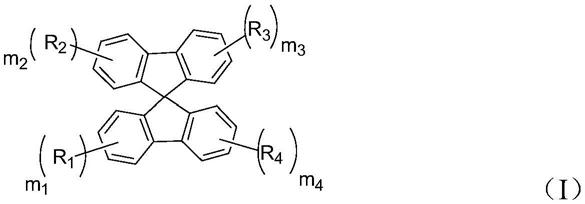
一种有机化合物,其具有如下式(i)所示结构特征:
[0007][0008]
其中,m1、m2、m3和m4分别独立的选自0或1,且m1和m2不同时为0,m3和m4不同时为0;
[0009]
r1和r2分别独立的选自含氮的供电子基团;
[0010]
r3和r4分别独立的选自
[0011]
本发明还提供一种有机发光二极管,采用具有式(1)所示结构特征的有机化合物作为蓝光发光材料:
[0012][0013]
其中,m1、m2、m3和m4分别独立的选自0或1,且m1和m2不同时为0,m3和m4不同时为0;
[0014]
r1和r2分别独立的选自含氮的供电子基团;
[0015]
r3和r4分别独立的选自
[0016]
与现有技术相比,本发明具有如下有益效果:
[0017]
本发明提供的有机化合物,以螺二芴为蓝光发射的核心,螺二芴本身为蓝光发射材料,结构为非平面结构,这种结构可削弱分子间堆积,从而降低浓度猝灭效应,而且螺二芴分子刚性大,稳定性好。另外,螺二芴右边部分结构(即r3和r4)为吸电子基团——四苯基吡嗪基,四苯基吡嗪本身可以进行深蓝光发射,且在固化工艺中分子结构的不易扭转,有利于固态发光;螺二芴左边部分结构(即r1和r2)采用含氮的供电子基团,与四苯基吡嗪配合形成双极性分子,有利于空穴和电子的传输。由此,具有本发明结构特征的有机化合物的优点如下:
[0018]
(1)该有机化合物作为蓝光材料,不需要主体材料,即不需要高能隙主体材料,减少不稳定性因素,从而有利于提高蓝光材料寿命,同时也可以用降低材料成本;
[0019]
(2)该有机化合物作为蓝光材料,制备的器件效率滚降低,寿命长。
[0020]
(3)该有机化合物易于制备得到,在电致发光、光伏电池、传感器等领域具有很大潜在应用。
附图说明
[0021]
图1为本发明一实施例所述的有机发光二极管的结构示意图。
具体实施方式
[0022]
以下结合具体实施例对本发明的有机化合物及包含其的有机发光二极管作进一步详细的说明。
[0023]
本发明实施例提供一种有机化合物,其具有如下式(i)所示结构特征:
[0024][0025]
其中,m1、m2、m3和m4分别独立的选自0或1,且m1和m2不同时为0,m3和m4不同时为0;
[0026]
r1和r2分别独立的选自含氮的供电子基团;
[0027]
r3和r4分别独立的选自(四苯基吡嗪)。
[0028]
在如上通式中,m3和m4分别独立的选自0或1,且m3和m4不同时为0。一个分子结构中,r3和r4的总取代数量不超过2,有利于基团的结构分布,减少分子间的平面堆积,进而减少浓度猝灭效应。
[0029]
在一些具体的实施例中,m1和m2中有一个为0;m3和m4中有一个为0。
[0030]
在一些具体的实施例中,该有机化合物具有如下式(i-1)或式(i-2)所示结构特征:
[0031][0032]
作为优选地,该有机化合物具有如下式(i-2)所示结构特征:
[0033][0034]
鉴于核心结构螺二芴具有两个扭曲的平面,相比较式(i-1),式(i-2)的螺二芴左右部分结构的扭曲分离程度更大,从而分子间的平面堆积更弱,猝灭作用更弱。
[0035]
作为优选地,r1和r2分别独立的选自三苯胺类及其衍生物和咔唑及其衍生物中的一种。该类型的r1和r2相比较其它的含氮供电子基团,具有更好的发光效率和稳定性。
[0036]
在一些具体的实施例中,r1和r2分别独立的选自如下式(i-a)、式(i-b1)、式(i-b2)和式(i-c)所示结构中的一种:
[0037][0038]
其中,r
11
和r
12
分别独立的选自h、或含1~5个碳原子的直链或支链烷基;
[0039][0040]
其中,r
21
和r
22
分别独立的选自h、或含1~5个碳原子的直链或支链烷基,ar为芳香基,m
22
为0或1;
[0041][0042]
其中,r
31
、r
32
和r
33
分别独立的选自h、或含1~5个碳原子的直链或支链烷基,ar为芳香基,m
31
为0、1或2,q为c、n、o、si或s。
[0043]
式(i-a)中,作为优选地,r
11
和r
12
分别独立的选自h、甲基、含4个碳原子的支链烷烃。更为优选地,r
11
和r
12
均为h。
[0044]
式(i-b1)和(i-b2)中,作为优选地,r
21
和r
22
分别独立的选自h、含4个碳原子的支链烷烃,ar为苯基,m
22
为0或1。
[0045]
式(i-c)中,作为优选地,r
31
、r
32
和r
33
分别独立的选自h或甲基,ar为苯基,m
31
为0、1或2,q为c、n、o、si或s。更为优选地,r
31
、r
32
和r
33
均为h,ar为芳香基,m
31
为2,q为c。
[0046]
在一些具体的实施例中,r1和r2分别独立的选自如下基团中的一种:
[0047][0048][0049]
在一些具体的实施例中,所述的有机化合物选自如下化合物m1-m34中的一种:
[0050]
[0051]
[0052]
[0053][0054]
在一些具体的实施例中,所述的有机化合物为一种蓝色发光材料。在具体应用时,该有机化合物可以单独作为蓝色发光材料使用,无需进行主体材料的掺杂,是一种非掺杂双极性蓝光材料。
[0055]
本发明的实施例还提供一种有机发光二极管,采用上述有机化合物作为蓝色发光材料。
[0056]
在一些具体的实施例中,该有机发光二极管包括空穴传输层、发光层和电子传输层,且所述空穴传输层、发光层和电子传输层中的至少一层为所述有机化合物。
[0057]
在一些具体的实施例中,所述发光层为所述蓝色发光材料。更为具体地,该有机发光二极管包括依次层叠设置的第一电极、空穴注入层(hil)、空穴传输层(htl)、发光层(eml)、电子传输层(etl)、电子注入层(eil)和第二电极;其中,所述发光层为所述有机化合物。
[0058]
在一些具体的实施例中,上述的有机化合物的制备方法包括如下步骤:
[0059]
在氮气氛围下,于有机溶剂中,采用式n-1所示的化合物依次与式m-3、m-4所示的化合物在催化剂作用下进行反应,即得。
[0060]
其中,x为br或i原子。d-b(oh)2式(m-4),其中,d代表所述含氮的供电子基团。
[0061]
在一些具体的实施例中,式(n-1)选自如下结构之一:
[0062][0063]
式(m-3)为如下结构:
[0064][0065]
d选自如下基团之一:
[0066][0067][0068]
更为具体地,本发明实施例中所公开的有机化合物m1~m34的通用合成路线如下所示:
[0069][0070]
参照上述反应路线:
[0071]
步骤1、中间体ii-1、ii-2的制备:
[0072]
于150ml的两口瓶中依次加入4mmol m-1(m-2)、4mmol m-3、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,加入搅拌磁子,进行抽真空换氮气操作,反复三次,使反应瓶内处于氮气氛围,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取2次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物ii-1(ii-2),最后室温真空干燥12h,称重,获得分离产率64%(71%)。1h nmr(500mhz,cdcl3),δ(tms,ppm):8.86(d,2h),8.03-8.08(m,7h),7.89(m,4h),7.78(t,2h),7.59-7.68(m,7h),7.23-7.41(m,11h)。{ii-2 1h nmr(500mhz,cdcl3),δ(tms,ppm):8.76(d,2h),8.01-8.07(m,7h),7.90(m,5h),7.78(d,2h),7.52-7.59(m,5h),7.28-7.41(m,12h)}。
[0073]
步骤2、最终产物iii-1、iii-2的制备:
[0074]
于150ml的两口瓶中依次加入2mmol ii-1或ii-2、2mmol d-b(oh)2(m-4)、0.1mmol四三苯基磷钯pd(pph3)4、4mmol碳酸钾k2co3,加入搅拌磁子,进行抽真空换氮气操作,反复三次,使反应瓶内处于氮气氛围,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取2次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物iii-1或iii-2。
[0075]
下面实施例1-4具体以化合物m1、化合物m9、化合物m16、化合物m30为例进行举例说明,参照上述步骤1、2很容易实现化合物m2~m8、m10~m15、m17~m29、m31~m34的合成。
[0076]
实施例1:化合物m1及其制备方法
[0077]
于150ml的两口瓶中依次加入2mmol ii-1、2mmol 4-硼酸三苯胺、0.1mmol四三苯基磷钯pd(pph3)4、4mmol碳酸钾k2co3,加入搅拌磁子,进行抽真空换氮气操作,反复三次,使反应瓶内处于氮气氛围,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取2次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物m1,最后室温真空干燥12h,称重。获得分离产率81%.1h nmr(500mhz,cdcl3),δ(tms,ppm):8.82(d,2h),8.03-8.09(m,8h),7.88(m,6h),7.78(d,2h),7.55-7.59(m,5h),7.24-7.32(m,18h),7.01-7.08(m,6h)。
[0078][0079]
实施例2:化合物m9及其制备方法
[0080]
于150ml的两口瓶中依次加入2mmol ii-1、2mmol 2-硼酸咔唑、0.1mmol四三苯基磷钯pd(pph3)4、4mmol碳酸钾k2co3,加入搅拌磁子,进行抽真空换氮气操作,反复三次,使反应瓶内处于氮气氛围,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取2次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物m9,最后室温真空干燥12h,称重。获得分离产率76%.1h nmr(500mhz,cdcl3),δ(tms,ppm):11.50(s,1h),8.83(d,2h),8.31(d,1h),8.19(m,1h),8.01-8.07(m,8h),7.89(m,7h),7.74-7.78(m,3h),7.59-7.63(m,7h),7.20-7.30(m,11h)。
[0081][0082]
实施例3:化合物m30及其制备方法
[0083]
于150ml的两口瓶中依次加入2mmol ii-2、2mmol(4-(10h-苯氧嗪-10-基)苯基)硼酸、0.1mmol四三苯基磷钯pd(pph3)4、4mmol碳酸钾k2co3,加入搅拌磁子,进行抽真空换氮气操作,反复三次,使反应瓶内处于氮气氛围,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取2次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物m30,最后室温真空干燥12h,称重。获得分离产率79%。1h nmr(500mhz,cdcl3),δ(tms,ppm):8.85(d,2h),8.03-8.07(m,8h),7.89(m,5h),7.76(d,2h),7.55-7.68(m,7h),7.28-7.37(m,13h),7.14(d,2h),6.96-7.01(m,6h)。
[0084][0085]
实施例4:化合物m16及其制备方法
[0086]
于150ml的两口瓶中依次加入2mmol ii-2、2mmol 2-甲基-9-苯基-9h-咔唑硼酸酯、0.1mmol四三苯基磷钯pd(pph3)4、4mmol碳酸钾k2co3,加入搅拌磁子,进行抽真空换氮气操作,反复三次,使反应瓶内处于氮气氛围,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取2次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物m16,最后室温真空干燥12h,称重。获得分离产率79%.1h nmr(500mhz,cdcl3),δ(tms,ppm):8.87(d,2h),8.55(d,1h),8.31(d,1h),7.89-8.09(m,15h),7.32-7.62(m,25h),7.16(t,1h)。
[0087][0088]
实施例5:有机发光二极管及其制备
[0089]
本实施例提供一种有机发光二极管元器件,其结构如图1所示,包含:第一电极(阳极)、在所述第一电极上形成的空穴注入层(hil)、在所述空穴注入层上形成空穴传输层(htl)、在所述空穴传输层上形成的发光层(eml)、在所述发光层上形成的电子传输层(etl)、在所述电子传输层上形成的电子注入层(eil)和覆盖在所述电子注入层上的第二电极(阴极);其中,所述发光层只包含上述实施例4的化合物m16。
[0090]
该有机发光二极管的制备方法包括如下步骤:
[0091]
(1)首先对ito基板按如下次序进行清洗:5%koh溶液超声15min、纯水超声15min、异丙醇超声15min、烘箱干燥1h;
[0092]
(2)然后将基板转移至uv-ozone设备进行表面处理15min,处理完后立即转移至手套箱中;
[0093]
(3)随后进行蒸镀成膜:依次制备空穴注入层、空穴传输层、发光层、电子传输层、电子注入层、第二电极;先抽真空至10-7
torr,然后缓慢提高电流值,使速率缓慢增加至待速率稳定后打开挡板进行蒸镀;
[0094]
(4)最后进行uv固化封装,再80℃烘烤20min即可。
[0095]
制备得到的ito/hil/htl/eml/etl/eil/阴极的多层有机发光二极管,结构为:
[0096]
ito/npb(10nm)/tcta(30nm)/m16(30nm)/tm3pypb(50nm)/naf(3.5nm)/al。
[0097]
其中,npb作为空穴注入层材料,tcta作为空穴传输层材料,m16作为发光层材料,tm3pypb作为电子传输层及空穴阻挡层材料,naf作为电子注入层材料,al作为阴极,该示例器件记作“m16器件”。
[0098]
参照该实施例的方法,采用化合物1至化合物34作为发光层材料制备有机发光二
极管,分别记作“m1器件”、“m2器件”,
……
,“m34器件”。
[0099]
对比例1
[0100]
本对比例以主客体材料体系为发光层,客体材料为经典的蓝色荧光材料2,5,8,11-四叔丁基芘tbpe:参照实施例5相同的方法制备有机发光二极管,记作“r1器件”,结构为:
[0101]
ito/npb(10nm)/tcta(30nm)/cbp:tbpe(10wt%)(30nm)/tm3pypb(50nm)/naf(3.5nm)/al。
[0102]
参照常规方法,对m1器件至m34器件及r1器件的最大外量子效率、效率滚降、寿命进行测试,结果参见表1。
[0103]
表1
[0104][0105]
注:效率滚降:电流效率从最高值到亮度10000nit对应的效率之间的变化率。
[0106]
由表1可知,采用本发明实施例的有机化合物制备的发光二极管效率滚降低(<10%)、寿命长。更进一步地,m1~m6采用三苯胺类基团作为含氮的供电子基团,m7~18采用咔唑类基团作为含氮的供电子基团,相较采用其它含氮的供电子基团,具有更低的效率滚
降和更长的器件寿命,其中采用咔唑类基团作为含氮的供电子基团的m7~18发光效率高、滚降低、寿命长,综合性能最优。另外,采用通式(i-2)的有机化合物较通式(i-1)滚降更低,说明猝灭作用更弱,同时发光效率更高、寿命更长,例如当采用的含氮的供电子基团均为咔唑类基团时,采用通式(i-2)的m13~18较采用通式(i-1)的m7~12的滚降更低、发光效率更高、且寿命更长。
[0107]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0108]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 1‑(2‑吡啶)‑9‑(2‑苯基乙基)‑β‑咔啉的硝酸铜配合物及其合成方法和应用与制造工艺
- 1‑(2‑吡啶)‑9‑(3‑苯基丙基)‑β‑咔啉的氯化铜配合物及其合成方法和应用与制造工艺
- 1‑(2‑吡啶)‑9‑(4‑甲基苄基)‑β‑咔啉的硝酸铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑丁基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑己基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑辛基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑壬基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑异戊基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 1‑(2‑吡啶)‑9‑(萘‑2‑甲基)‑β‑咔啉的氯化铜配合物及其合成方法和应用与制造工艺
- 1‑(2‑吡啶)‑9‑(2‑乙氧基乙基)‑β‑咔啉的氯化铜配合物及合成方法和应用与制造工艺