一种用于合成布瓦西坦的中间体及其制备方法与流程
[0001]
本发明涉及布瓦西坦合成技术领域,更具体地,涉及中间体(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i),以及其合成技术领域。
[0002]
背景技术:
[0003]
布瓦西坦(brivaracetam)是ucb公司开发的新一代抗癫痫药物,2016年2月,美国fda批准布瓦西坦上市,用于治疗16岁及以上部分发作型的癫痫患者,辅助治疗伴随或不伴随继发全身性发作。布瓦西坦(s)-2-((r)-2-氧代-4-丙基吡咯啉-1-基)丁酰胺的结构如下所示。
[0004]
由于布瓦西坦分子中有两个手性中心,对应四个不同的异构体,这给布瓦西坦的合成带来了一定的难度。目前合成布瓦西坦的方法主要有以下报道:最早的方法是ucb开发的,以正戊醛为原料,先与乙醛酸环合,再与l-氨基丁酰胺反应,得到的产物氢化,生成一对非对映异构体,再通过柱层析分离纯化得到产物布瓦西坦,如专利cn1208319c、cn1882535b所述。该方法步骤虽短,但最后一步只能柱层析分离,成本高而且不适合大量生产,限制了其应用。
[0005]
cn107663185a报道了以(r)-4-丙基二氢呋喃-2(3h)-酮为中间体与l-氨基丁酰胺反应得到布瓦西坦。但是手性的(r)-4-丙基二氢呋喃-2(3h)-酮合成路线很长,涉及到金属试剂反应,而且它与l-氨基丁酰胺也需要多步转化才能得到目标产物。这使整个合成方案路线长,操作繁琐。
[0006]
cn106748950a报道了以差向异构体混合物酸为中间体,通过与r-苯乙胺的成盐纯化的方法得到单一异构体酸,再转化成布瓦西坦的方法。该方法相对比较简洁,实用性较强。
技术实现要素:
[0007]
为了克克服现有的布瓦西坦制备方法中存在的成本高、步骤冗长、操作繁琐、不适合工业化生产的缺陷,本发明提出了一种用于合成布瓦西坦中间体(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)。
[0008]
该化合物(i)包含(2s)-2-(((r)-5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)-r,或(2s)-2-(((s)-5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)-s,或者任意比例的(i)-r与(i)-s的混合物。
[0009]
化合物(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)具体有以下谱图上的特征:1h nmr (500 mhz, chloroform-d) δ 6.45 (s, 1h), 5.89 (s, 1h), 5.85 (s, 1h), 5.57 (s, 1h), 3.42 (s, 1h), 2.39 (s, 3h), 1.82 (dq, j = 14.4, 7.4, 6.9 hz, 1h), 1.74 (dt, j = 14.2, 7.2 hz, 1h), 1.67 (dd, j = 14.6, 7.2 hz, 2h), 1.02 (dt, j = 11.3, 7.4 hz, 6h).ms (esi) m/z =227 (m
+
+1).ftir(kbr)ν
max
: 3412,2975, 1727, 1637, 1436, 1317, 930, 911, 875, 742 cm-1
,其中1727cm-1
和1637cm-1
处,分别是内酯的羰基吸收峰和酰胺的羰基吸收峰。
[0010]
本发明提出了一种制备(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)的方法,包括将5-羟基-4-丙基呋喃-2(5h)-酮(iii)与l-氨基丁酰胺或其可接受的盐(ii)在醇类溶剂中反应,得到式(i)的化合物。
[0011]
其中,l-氨基丁酰胺可接受的盐(ii)包含盐酸盐、醋酸盐、酒石酸盐;其中,醇类溶剂包含甲醇、乙醇、异丙醇。
[0012]
较佳地,制备(2s)-2-(((r)-5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)的反应温度为20~50
o
c,时间为2~4小时。
[0013]
本发明还提出一种制备3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)或其可接受的盐的方法,包括在溶剂中以及催化剂的作用下,将(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)与氢气进行如下所示的氢化还原反应,得如式(iv)所示化合物;
其中,氢化还原反应的催化剂包括钯碳催化剂;其中,溶剂包括水、c
1~4
醇,或者任意比例的水和c
1~4
醇的混合物。
[0014]
其中,3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)为(r)-3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)-r、(s)-3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)-s,或其任意比例的(iv)-r和(iv)-s的混合物;其中,3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)可接受的盐包含柠檬酸盐、草酸盐和马来酸盐。
[0015]
较佳地,氢化反应的反应温度为10~50
o
c,压力1~50bar,反应时间2~6小时。
具体实施方式
[0016]
为更好的理解本发明的内容,下面结合具体实施例作进一步说明。应理解,下列具体实施例仅仅用于说明本发明,而不是对本发明的限制。
[0017]
实施例1:5-羟基-4-丙基呋喃-2(5h)-酮(iii)的制备将193kg吗啡啉抽入到1000l反应釜中,降温至10℃以下,缓慢加入水中;滴加50%乙醛酸水溶液450kg,控温不超过30℃。滴完后,25~30℃保温反应5小时;然后滴加182kg正戊醛,40~45℃保温反应18小时。
[0018]
降温至20℃,缓慢滴加浓盐酸,25~30℃保温反应4小时,再将反应液转至2000l反应釜中,缓慢加入碳酸钠固体调ph至4~5,用乙酸乙酯萃取三次,合并有机相后,用饱和食盐水洗涤,无水硫酸钠干燥、抽滤、减压浓缩,得300kg棕色油状物5-羟基-4-丙基呋喃-2(5h)-酮(iii)43.9g,含量80%,折合收率95.8%。1h nmr (400 mhz, chloroform-d) δ 0.93-1.00 (t, 3h), 1.56-1.67 (q, 2h), 2.31-2.43 (q, 2h), 5.81 (s, 1 h), 6.02 (s, 1 h)
.ms (esi) m/z =143 (m
+
+1).实施例2:(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)的制备往198kg的l-氨基丁酰胺盐酸盐中加入甲醇1600kg,通氨气进行游离,直至体系ph值为9~10,且ph值不变化为止。过滤除去盐,滤液浓缩至500l待用。
[0019]
将300kg的5-羟基-4-丙基呋喃-2(5h)-酮(iii)分批加入上述500l氨基丁酰胺的溶液中,控温20~30
o
c反应2小时。反应完成后过滤去除盐,滤液缓慢降温至0~5
o
c析晶,离心,得固体湿重320kg,固体再用乙酸乙酯重结晶,得245kg白色固体(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i),收率79.8%。1h nmr (500 mhz, chloroform-d) δ 6.45 (s, 1h), 5.89 (s, 1h), 5.85 (s, 1h), 5.57 (s, 1h), 3.42 (s, 1h), 2.39 (s, 3h), 1.82 (dq, j = 14.4, 7.4, 6.9 hz, 1h), 1.74 (dt, j = 14.2, 7.2 hz, 1h), 1.67 (dd, j = 14.6, 7.2 hz, 2h), 1.02 (dt, j = 11.3, 7.4 hz, 6h).ms (esi) m/z =227 (m
+
+1).ftir (kbr)ν
max
: 3412,2975, 1727, 1637, 1436, 1317, 930, 911, 875, 742 cm-1
.实施例3:(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)的制备往116kg的l-氨基丁酰胺醋酸盐中加入乙醇800kg,通氨气进行游离,直至体系ph值为9~10,且ph值不变化为止。过滤除去盐,滤液浓缩至250l待用。
[0020]
将150kg的5-羟基-4-丙基呋喃-2(5h)-酮(iii) 98.4g分批加入上述250l氨基丁酰胺的溶液中,控温30~40
o
c反应3小时。反应完成后过滤去除盐,滤液缓慢降温至0~5
o
c析晶,离心,得固体湿重167kg,固体再用乙酸乙酯重结晶,得130kg白色固体(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i),收率80.3%。谱图见实施例2。
[0021]
实施例4:(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i)的制备
往241kg的l-氨基丁酰胺l-酒石酸盐中加入异丙醇1000kg,通氨气进行游离,直至体系ph值为9~10,且ph值不变化为止。过滤除去盐,滤液浓缩至350l待用。
[0022]
将200kg的5-羟基-4-丙基呋喃-2(5h)-酮(iii)分批加入上述350l氨基丁酰胺的溶液中,控温40~50
o
c反应2小时。反应完成后过滤去除盐,滤液缓慢降温至0~5
o
c析晶,离心,得固体湿重350kg,固体再用乙酸乙酯重结晶,得155kg白色固体(2s)-2-((5-氧化-3-丙基-2,5-二氢呋喃-2-基)氨基)丁酰胺(i),收率71.7%。谱图见实施例2。
[0023]
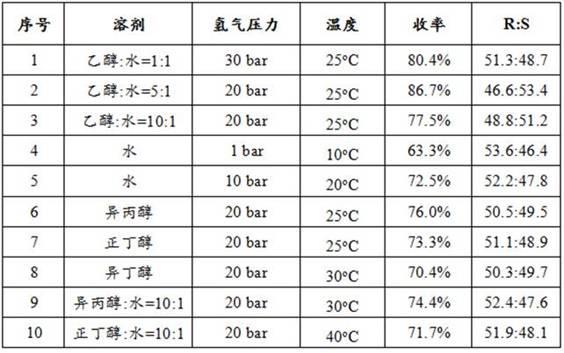
实施例5:3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)的制备将100g的(2s)-2-(2-羟基-5-氧代-3-丙基-2,5-二氢-1h-吡咯-1-基)丁酰胺(ii)加入到150ml水与150ml乙醇的混合溶剂中,再加入5%的pd/c7.5g, 搅拌均匀,氮气置换。通氢气至30bar,常温搅拌反应3小时。反应完后,过滤去钯碳,旋干滤液,得到81.8g淡黄色油状物3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv),收率80.4%。hplc测得化合物(iv)中(r)-3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)-r与(s)-3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)-s的比例,即(iv)-r:(iv)-s=51.3:48.7%。1h nmr (400 mhz, dmso-d6) δ 7.34 (s, 1h), 7.01 (s, 1h), 2.96
ꢀ–ꢀ
2.79 (m, 1h), 2.56 (dd, j = 11.6, 4.9 hz, 1h), 2.40 (d, j = 4.8 hz, 1h), 2.29 (ddt, j = 28.6, 20.1, 10.3 hz, 1h), 2.13 (ddd, j = 21.1, 11.9, 5.2 hz, 1h), 1.93
ꢀ–ꢀ
1.74 (m, 1h), 1.49 (tt, j = 13.6, 6.5 hz, 2h), 1.41
ꢀ–ꢀ
1.07 (m, 4h), 0.82 (dt, j = 29.2, 6.6 hz, 6h).ms (esi) m/z =231 (m
+
+1).重复上述还原反应操作,采用不同的溶剂、温度和氢气压力,反应完成后,分离计算收率,测(iv)-r与(iv)-s的比例,实验结果如下表所示:
实施例6:3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)草酸盐的制备将200g的(2s)-2-(2-羟基-5-氧代-3-丙基-2,5-二氢-1h-吡咯-1-基)丁酰胺(ii)加入到50ml水与400ml异丙醇的混合溶剂中,再加入5%的pd/c 15.0g, 搅拌均匀,氮气置换。通氢气至30bar,常温搅拌反应3小时。反应完后,过滤去钯碳,往得到的滤液加入草酸160g,搅拌成盐,再加入800ml甲基叔丁基醚,搅拌25~30℃析晶1-2小时,抽滤,用甲基叔丁基醚淋洗,干燥,得150g白色固体3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)草酸盐,hplc测得化合物(iv)中(iv)-r:(iv)-s=68.7:31.3。
[0024]
将150g白色固体3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)草酸盐溶于加入正丁醇和水中,加入甲基叔丁基醚析晶,得到的固体再如此重结晶一次,最后得到99g白色固体(r)-3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)-r草酸盐,其中(iv)-r:(iv)-s=99.5:0.5,收率33.5%。1h nmr (400 mhz, dmso-d6) δ 8.05 (s, 1h), 7.65 (s, 1h), 3.63 (s, 1h), 2.85 (t, j = 9.5 hz, 1h), 2.78
ꢀ–ꢀ
2.61 (m, 1h), 2.43 (dd, j = 16.5, 5.8 hz, 1h), 2.25 (dd, j = 16.6, 6.6 hz, 1h), 2.10 (s, 1h), 1.78 (ddt, j = 30.1, 14.9, 7.5 hz, 2h), 1.57
ꢀ–ꢀ
1.11 (m, 4h), 0.88 (dt, j = 13.4, 7.1 hz, 6h).ms (esi) m/z =231 (m
+
+1).
重复上述还原反应操作,采用不同的有机酸成盐,分离计算收率,测(iv)-r与(iv)-s的比例,实验结果如下表所示:实施例7:布瓦西坦的制备将80g异构体比例为(iv)-r:(iv)-s==99.5:0.5的(r)-3-((((s)-1-氨基-1-氧代丁-2-基)氨基)甲基)己酸(iv)-r草酸盐溶于含有500ml异丙醇中,加热至回流反应6小时后降温,减压浓缩;得到的浓缩液用10%的碳酸钠水溶液调节体系ph至8~9,再用二氯甲烷萃取,干燥,浓缩,得粗品52g。
[0025]
将粗品溶于50ml乙酸乙酯,滴加正庚烷250ml,室温搅拌结晶1小时,抽滤,正庚烷淋洗,烘干得布瓦西坦(i)47g,异构体比例sr:ss=99.5:0.5,收率:89.3%。1h nmr (400 mhz, cdcl3) δ 6.53 (s, 1h), 5.89 (s, 1h), 4.43 (dd, j = 9.0, 6.8 hz, 1h), 3.44 (dd, j = 9.8, 7.9 hz, 1h), 3.01 (dd, j = 9.8, 7.1 hz, 1h), 2.52 (dd, j = 16.7, 8.6 hz, 1h), 2.35
ꢀ–ꢀ
2.18 (m, 1h), 2.04 (dd, j = 16.7, 8.1 hz, 1h), 1.87 (dd, j = 14.2, 7.1 hz, 1h), 1.74
ꢀ–ꢀ
1.47 (m, 1h), 1.43
ꢀ–ꢀ
1.33 (m, 2h), 1.27 (ddd, j = 11.4, 8.2, 5.8 hz, 2h), 0.85 (dd, j = 13.6, 7.3 hz, 6h). ms (esi) m/z =213 (m+h)
+
.在此说明书中,本发明已参照其特定的实施例作了描述。但是,很显然仍可以作出各种修改和变换而不背离本发明的精神和范围。因此,说明书应被认为是说明性的而非限制性的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1