若干硝基化合物的硅烷化学还原制备胺基产物及H-酸的新环保生产工艺的制作方法
若干硝基化合物的硅烷化学还原制备胺基产物及h-酸的新环保生产工艺
【技术领域】
1.本发明涉及精细化学品新材料领域,涉及系列硝基化合物的还原反应,特别涉及间硝基苯胺、间苯二胺、5-氨基邻甲酚、2-甲基对苯二胺、1/2-萘胺、h-酸胺、2,4,6-三甲基间苯二胺等若干特定重要胺类化合物自其相应单或双硝基化合物前体进行硅烷化学还原的温和高效,环境友好,且经济性制备新工艺技术,以及由此衍生的h-酸的新环保生产工艺技术。
背景技术:
2.本发明关注如下特定单或双胺官能型化合物的新型环境友好和低成本制备工艺技术。这些化合物包括了间硝基苯胺、间苯二胺、5-氨基邻甲酚、2-甲基对苯二胺、1/2-萘胺、h-酸胺、和2,4,6-三甲基间苯二胺等在染料,颜料,农药、医药、彩色显影剂、塑料、合成纤维及功能助剂产业领域有广泛应用的重要中间体或终端产品。
[0003][0004]
当前上述芳香胺类化合物的工业生产主要利用硝基还原法,根据还原剂的不同又可以细分为金属还原法,含硫化合物还原法,催化氢化法,以及水合肼还原法。金属还原法常用锌/铁等活泼金属在酸性条件下还原芳香硝基化合物,例如间二硝基苯经铁粉还原制备间硝基苯胺(中国申请cn100494163)。该方法具有产品选择性好、设备要求低、工艺简洁等优点,是目前我国生产芳香胺类化合物的主要方法。但该方法存在严重的金属污染,含盐废水和含有机物的铁泥废渣处理等问题。含硫化合物还原法选用多硫化合物来还原芳香硝基化合物,例如间二硝基苯经多硫化钠部分还原制备间硝基苯胺(徐克勋,精细化工原料及中间体手册,北京:化学工业出版社,2001,5-531-532)。该方法反应温和,工艺简单,对设备要求低,适用范围广,但产率低,且生产中产生大量含硫废水。催化氢化法需要使用价格昂贵的铂/钯/镍等催化剂(例如间苯二胺合成中相应专利:中国申请cn10132357,中国申请cn108164425,中国申请cn10343305),存在贵金属催化剂回收以及失活的问题,且高压条件和易燃易爆的氢气的使用也给设备提出苛刻的要求。水合肼还原法(例如间二硝基苯水合肼还原制备间苯二胺,m.s.thakur,o.s.nayal,r.rana,m.kumar,s.sharma,n.kumar,s.k.maurya.new j.chem.2018,42,1373.),具有反应条件温和、还原收率高、不产生废渣废水、后处理简单等特点,但还原时需要载体或催化剂的存在(如贵金属有机催化剂、pd/c、
fecl3/c、raney ni等),面临催化剂回收,成本偏高等问题。专利cn104822642批露了特定氢硅烷对硝基化合物的还原,但是尚不清楚其适用性和实用性,特别是针对本发明诉求的特定化学结构和应用功能的单或双官能硝基化合物的还原。
[0005]
由以上当前的工艺技术状况总结可知,仍然亟需发展简单温和,高效安全,特别是绿色环保且具备经济竞争力的新型硝基还原工艺来生产特定芳胺类精细化工产品,以应对市场日益增长的需求。
技术实现要素:
[0006]
本项申请现已发现,对所述感兴趣的特定单或双官能芳胺类化合物,可以经由方程式(i-viii)描述的反应技术,自其相应的芳香硝基化合物前体和硅烷还原剂【si-re】在适当的反应条件conditions下作用实现制备。conditions是指有机碱、热、光、真空或压力、溶剂、添加剂等条件中的任意一种,或是上述条件中的任意两种或两种以上因素,的联合使用。【si-re】是三氯硅烷(hsicl3),和/或“三氯硅烷-叔胺配合物”(分子式通式为hcl3si
→
nr1r2r3);这里r1,r2,r3分别独立地是c
1-c
24
的直链或支链的烷基,或c
4-c
24
的取代或未取代的(杂)芳基。当q1=no2时,q2=nh2;当q1=so3h或so3阴离子时,q2=q1。
[0007]
本工艺所述有机碱是含有n和/或p的脂肪族或芳香族胺或叔膦类物质;示例性而非限定性的例子是三乙胺、二乙胺、三丙胺、三丁胺、二异丙基胺、二异丙基乙基胺、n,n-二甲基苯胺、n-甲基苯胺、n,n-四甲基乙二胺、4-n,n-二甲基吡啶(dmap)、dbu、dbn、苯胺、苄胺、环己胺、吡咯烷、吡啶、二乙烯三胺、三乙烯四胺、五乙烯六胺、n(ch2chr)3p、verkade碱。
[0008][0009]
有机碱和三氯硅烷分别独立地和原料的摩尔比值在(0.001-1000)/1,优选(0.01-100)/1,更优选(1-50)/1。
[0010]
本工艺所涉及的反应“溶剂”选自含有1-24个碳的取代或非取代的芳香烃,直链或支链的脂肪烃,酰胺,醚,酯,碳酸酯,酮,腈,羧酸,水,胺,离子液,超临界二氧化碳,或上述任意二者或二者以上类型溶剂组成的混合溶剂;优先的溶剂是二氯甲烷,二氯乙烷,氯仿,四氯化碳,苯,甲苯,二甲苯,三甲苯,四甲苯,乙苯,二乙苯,氯苯,二氯苯,苯甲醚,硝基苯,庚烷,己烷,石油醚,二氧六环,四氢呋喃,甲基叔丁基醚,乙二醇二甲醚,双缩乙二醇二甲
醚,三缩乙二醇二甲醚,丙二醇甲醚醋酸酯,三乙胺,三丁胺,二甲基异丙胺,吡啶,n,n-四甲基乙二胺,n-烷基吗啉,n-烷基吡咯,n,n-二甲基甲酰胺,hmpa,甲酰吗啉,n,n-二乙基甲酰胺,n-甲基吡咯烷酮,碳酸二甲酯,碳酸二乙酯,或上述任意二者或二者以上溶剂组成的混合溶剂;本反应工艺各模块也可以不使用溶剂,而直接以其液态反应物或溶解体或加热熔融的液态物料体系为介质进行反应。
[0011]
本工艺涉及的反应“温度”选自-50-450摄氏度之间。
[0012]
本工艺涉及的反应“真空或压力”选自0.01-1
×
106帕斯卡之间。
[0013]
本工艺所涉及的反应“添加剂”涵盖反应促进剂,增效剂,催化剂,解络剂,酸或碱中和剂,和/或功能助剂,其是路易斯酸或路易斯碱型(lewis acids/bases)单质或化合物,优选的是碱金属,碱土金属,主族金属,或过渡金属的氟化物,氯化物,溴化物,碘化物,氧化物,氢氧化物,硫化物,烷氧化物,烷基或芳基金属化合物;或碱金属,碱土金属,主族金属,或过渡金属的碳酸盐,碳酸氢盐,亚硫酸盐,硫酸氢盐,磺酸盐,或羧酸盐;或碱金属,碱土金属,或过渡金属的一元或多元有机磷,有机胺,羟基,酮羰基,酯羰基,或羧酸配体的配合物;或四烷基卤化铵,四烷基氢氧化铵,或离子液体(ionic liquids);或无机质子酸(包括但不限于硫酸、盐酸、磷酸、硝酸、醋酸),有机羧酸,有机磺酸,杂多酸,分子筛,沸石,硅藻土,蒙拓土,高岭土;或硼,硅,磷元素的氟化物,氯化物,溴化物,碘化物,氧化物,氢氧化物,硫化物,烷氧化物;或满足上述定义“添加剂”中任意二者或二者以上的混合物或其联合使用。以反应原材料摩尔当量为基准,所述“添加剂”的使用量可以是催化量,等当量,或过当量。
[0014]
本发明同时披露一种h-酸的新环保生产工艺技术,其包含的反应序列如下:第一步,自萘和发烟硫酸和/或三氧化硫进行磺化反应得到1,3,6-三磺酸萘;第二步,1,3,6-三磺酸萘和硝酸进行硝化反应得到1-硝基-3,6,8-三磺酸基萘;第三步,1-硝基-3,6,8-三磺酸基萘和本发明批露的硅烷还原剂【si-re】在适当的反应条件conditions下作用得到1-胺基-3,6,8-三磺酸基萘;第四步,1-胺基-3,6,8-三磺酸基萘发生碱熔反应得到h-酸目标产物。
[0015][0016]
本发明同时披露另一种h-酸的新环保生产工艺技术,其包含的反应序列如下:第一步,自萘和发烟硫酸和/或三氧化硫进行磺化反应得到2,7-二磺酸萘;第二步,2,7-二磺酸萘和硝酸进行硝化反应得到1,8-二硝基-3,6-二磺酸基萘;第三步,1,8-二硝基-3,6-二磺酸基萘和本发明批露的硅烷还原剂【si-re】在适当的反应条件conditions下作用得到1,8-二胺基-3,6-二磺酸基萘;第四步,1,8-二胺基-3,6-二磺酸基萘发生酸性水合反应得到h-酸目标产物。
[0017][0018]
三氯硅烷是极为价廉易得的工业原材料,同时,其反应副产物经过水解处理后不产生有毒有害物质。因此,该披露工艺赋予了上述目标产物工业生产极具绿色环保和低成本经济竞争力的应用特征。
【具体实施方式】
[0019]
下面结合具体实施例进一步说明本发明要旨:
[0020]
实施例一:间硝基苯胺的合成
[0021][0022]
在氮气保护下,将16.8克间二硝基苯和300毫升乙腈加入到1升三口瓶中,室温下加入64.5克二异丙基乙基胺,后冰浴下滴加40.6克三氯硅烷于100毫升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应2小时后。随后反应体系加入200ml饱和碳酸氢钠溶液,二氯甲烷萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到淡黄色固体12.6克,气相色谱-质谱(gc-ms)分析表明含有95%的间硝基苯胺。硅胶柱色谱分离得到12.7克的间硝基苯胺(92%收率)。叔胺助剂即二异丙基乙基胺可以回收套用。
[0023]
重复该反应,但是还原剂使用80克的“三氯硅烷-二异丙基乙基胺配合物”【hcl3si
→
net(
i
pr)2】,得到12.1克间硝基苯胺目标产物。
[0024]
实施例二:间硝基苯胺的合成
[0025][0026]
在氮气保护下,将168克间二硝基苯和3升乙腈加入到10升三口瓶中,室温下加入925克三丁基胺,后冰浴下滴加406克三氯硅烷于1升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入2升饱和碳酸氢钠溶液,二氯甲烷萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到黄色固体123克。黄色固体经过石油醚/乙酸乙酯重结晶得淡黄色晶体122.8克,收率89%。核磁数据:1h-nmr(400mhz,cdcl3):δ=7.59(dd,j=8.0,3.6hz,1h),7.51(t,j=3.6hz,1h),7.30(d,j=8.0hz,1h),6.98(dd,j=8.0,3.6hz,1h),4.06(s,br,2h);
13
c-nmr(100mhz,cdcl3):δ=149.4,147.5,129.9,120.7,113.1,109.0.
[0027]
实施例三:间苯二胺的合成
[0028][0029]
在氮气保护下,将16.8克间二硝基苯和300毫升乙腈加入到1升三口瓶中,室温下加入90.3克二异丙基乙基胺,后冰浴下滴加67.7克三氯硅烷于100毫升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应2小时后。随后反应体系加入200ml饱和碳酸氢钠溶液,二氯甲烷萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到白色固体9.4克。气相色谱-质谱(gc-ms)分析表明含有96%的间苯二胺。硅胶柱色谱分离得到10.2克的间苯二胺(94%收率)。
[0030]
实施例四:间苯二胺的合成
[0031][0032]
在氮气保护下,将168克间二硝基苯和3升乙腈加入到10升三口瓶中,室温下加入707克三乙胺,后冰浴下滴加677克三氯硅烷于1升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入2升饱和碳酸氢钠溶液,二氯甲烷萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到淡黄色固体110克。黄色固体经过石油醚/乙酸乙酯重结晶得白色晶体95克,收率88%。核磁数据:1h-nmr(400mhz,cdcl3):δ=6.97(t,j=7.8hz,1h),6.14(dd,j=7.8,1.9hz,2h),6.03(s,1h),3.60(s,br,4h);
13
c-nmr(100mhz,cdcl3):δ=147.6,130.2,106.0,102.0.
[0033]
重复该反应,但是还原剂使用1330克的“三氯硅烷-二异丙基乙基胺配合物”【hcl3si
→
net(
i
pr)2】,得到99.3克间苯二胺目标产物。
[0034]
实施例五:5-氨基邻甲酚的合成
[0035][0036]
在氮气保护下,将7.6克5-硝基邻甲酚和100毫升二氯甲烷加入到500毫升三口瓶中,室温下加入45.1克二异丙基乙基胺,后冰浴下滴加33.8克三氯硅烷于100毫升的二氯甲烷溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入250毫升饱和碳酸氢钠溶液,二氯甲烷萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到黄色固体6.0克。黄色固体经过乙醇重结晶、打浆得米白色晶体5.7克,收率92%。核磁数据:1h-nmr(400mhz,cdcl3):δ=6.88(d,j=8.0hz,1h),6.21(dd,j=8.0,2.3hz,1h),6.18(d,j=2.3hz,1h),4.64(s,br,1h),3.50(s,br,1h),2.13(s,3h),1.59(s,br,1h).
[0037]
实施例六:2-甲基对苯二胺的合成
[0038][0039]
在氮气保护下,将15.2克4-硝基邻甲苯胺和300毫升二氯甲烷加入到1升三口瓶中,室温下加入64.5克二异丙基乙基胺,后冰浴下滴加40.6克三氯硅烷于200毫升的二氯甲烷溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入500毫升饱和碳酸氢钠溶液,二氯甲烷萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到黄色固体11.5克。黄色固体经过石油醚/乙酸乙酯重结晶浅黄色晶体10克,收率82%。核磁数据:1h-nmr(400mhz,cdcl3):δ=6.54(d,j=8.1hz,1h),6.50(d,j=2.6hz,1h),6.45(dd,j=8.1,2.6hz,1h),3.26(s,br,4h),2.12(s,3h).
[0040]
实施例七:2-甲基对苯二胺的合成
[0041][0042]
在氮气保护下,将15.2克3-甲基-4-硝基苯胺和300毫升二氯甲烷加入到1升三口瓶中,室温下加入64.5克二异丙基乙基胺,后冰浴下滴加40.6克三氯硅烷于200毫升的二氯甲烷溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入500毫升饱和碳酸氢钠溶液,dcm萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到黄色固体11.8克。黄色固体经过石油醚/乙酸乙酯重结晶得黄色晶体11克,收率90%。
[0043]
实施例八:1-萘胺的合成
[0044][0045]
在氮气保护下,将8.6克1-硝基萘和100毫升乙腈加入到500毫升三口瓶中,室温下加入32.2克二异丙基乙基胺,后冰浴下滴加20.3克三氯硅烷于100毫升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入250毫升饱和碳酸氢钠溶液,乙酸乙酯萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到黄色固体7.3克。黄色固体经过石油醚/乙酸乙酯重结晶得白色针状晶体7.5克,收率91%。核磁数据:1h-nmr(400mhz,cdcl3):δ=7.71-7.77(m,2h),7.36-7.43(m,2h),7.22-7.29(m,2h)6.68(dd,j=7.2,1.2hz,1h),4.01(s,br,2h).
[0046]
实施例九:2-萘胺的合成
[0047][0048]
在氮气保护下,将17.3克2-硝基萘和300毫升乙腈加入到1升三口瓶中,室温下加入64.5克二异丙基乙基胺,后冰浴下滴加40.6克三氯硅烷于200毫升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入500毫升饱和碳酸氢钠溶液,乙酸乙
酯萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到黄色固体16.9克。黄色固体经过石油醚/乙酸乙酯重结晶得白色晶体13.3克,收率93%。核磁数据:1h-nmr(400mhz,cdcl3):δ=7.71(d,j=8.4hz,1h),7.68(d,j=8.4hz,1h),7.61(d,j=8.0hz,1h),7.39(t,j=7.2hz,1h),7.25(t,j=6.8hz,1h),7.01(s,1h),6.97(dd,j=8.8hz,2.0hz,1h),3.86(s,2h).
[0049]
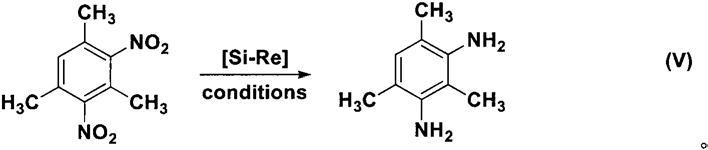
实施例十:2,4,6-三甲基-1,3-苯二胺的合成
[0050][0051]
在氮气保护下,将4.2克1,3-二硝基-2,4,6-三甲基苯和100毫升乙腈加入到500毫升三口瓶中,室温下加入18.1克二异丙基乙基胺,后冰浴下滴加13.5克三氯硅烷于100毫升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入500毫升饱和碳酸氢钠溶液,乙酸乙酯萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到黄色固体3.2克。黄色固体经过石油醚/乙酸乙酯重结晶得淡黄色晶体2.9克,收率95%。
[0052]
实施例十一:1,8-二氨基-3,6-二磺酸的合成
[0053][0054]
在氮气保护下,将3.5克1,8-二硝基-3,6-二磺酸和100毫升乙腈加入到500毫升三口瓶中,室温下加入14.3克二异丙基乙基胺,后冰浴下滴加8.3克三氯硅烷于30毫升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应12小时后。随后反应体系加入150毫升饱和碳酸氢钠溶液,乙酸乙酯萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后浓缩得到淡黄色固体3.5克。淡黄色固体经过石油醚/乙酸乙酯重结晶得晶体2.6克,收率88%。
[0055]
实施例十二:h-酸的合成
[0056][0057]
在氮气保护下,自936克萘出发,参考实施例十一的关键步骤实施上述反应序列,最终以74%的全程总收率得到1725.5克h-酸产品。
[0058]
实施例十三:1-胺基-3,6,8-三磺酸的合成
[0059][0060]
在氮气保护下,将30.6克1-硝基-3,6,8-三磺酸基萘和450毫升乙腈加入到1升三口瓶中,室温下加入66.6克二异丙基乙基胺,后冰浴下滴加46.9克三氯硅烷于100毫升的乙腈溶液,加毕由0℃缓缓升至室温搅拌反应3小时后。随后反应体系加入300ml饱和碳酸氢钠溶液,二氯甲烷萃取,有机相用饱和食盐水洗涤、mgso4干燥、活性炭脱色处理后,乙醇结晶纯化得到25.4克1-胺基-3,6,8-三磺酸产品,收率90%。
[0061]
实施例十四:h-酸的合成
[0062][0063]
在氮气保护下,自877克萘出发,参考实施例十三的关键步骤实施上述反应序列,最终以68%的全程总收率得到1485.7克h-酸产品。
[0064]
需要强调的是,上述实施例仅仅为示例性而非限定性说明,基于本项申请披露,任何从业技术人员所通常可能采用的反应条件或参数等调整或变动均不会偏离本发明的要旨,本专利的保护范围应以相关的权利书记载条目为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1