一种2,4-二氟-3-硝基苯甲酸的制备方法与流程
本发明涉及一种2,4-二氟-3-硝基苯甲酸的制备方法,属于医药技术(有机合成)领域。
背景技术:
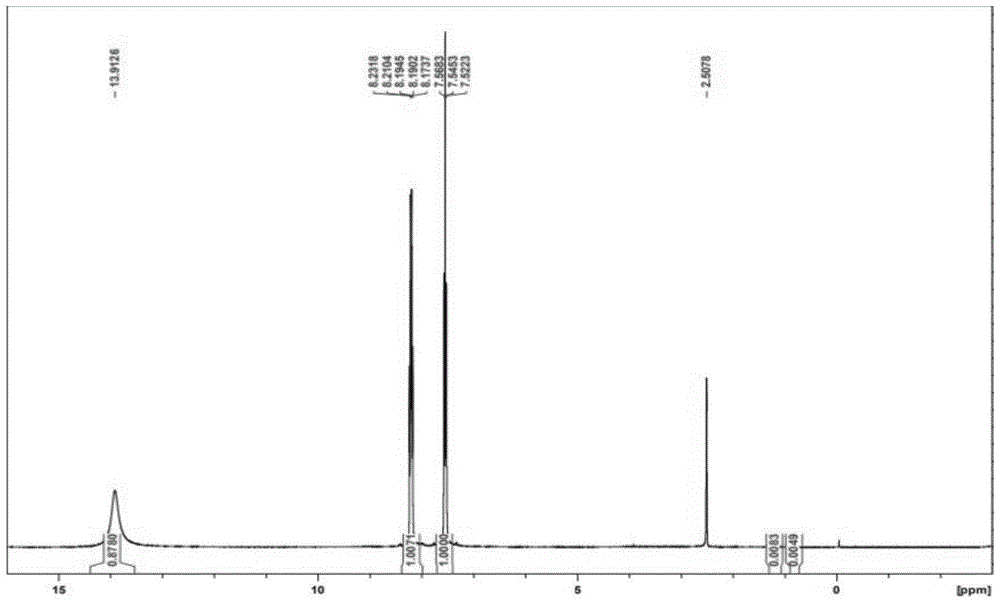
:2,4-二氟-3-硝基苯甲酸,cas登录号:1425174-31-7,英文名称是:2,4-difluoro-3-nitrobenzoicacid。结构式如下:2,4-二氟-3-硝基苯甲酸结构中含有两个氟原子、硝基和羧基,可以用于构建新药分子结构,是一种新颖的医药合成中间体。2,4-二氟-3-硝基苯甲酸可以用于合成药物候选化合物,例如:heparegenixgmbh+pctint.appl.(2019),wo2019243315a1[de]tricyclicproteinkinaseinhibitorsforpromotingliverregenerationorreducingorpreventinghepatocytedeath,筛选测试具有良好生物活性的药物,如下列结构式i和结构式ii。例如:pctint.appl.(2012),wo2012171488a1提供一种具有抗肿瘤及抗炎活性的新型香豆素衍生物或其药学可接受的盐或溶剂化物、包含该衍生物的药物组合物,如下列结构式iii具有良好的抗肿瘤活性。例如:pctint.appl.(2019),wo2019053426a1发明涉及新的化合物和成分,以及它们在治疗高血糖和高血糖特征疾病(如ii型糖尿病)中的应用。如下列结构式iv和式v都显示了良好的降糖生物活性。以上系列化合物,都可以以2,4-二氟-3-硝基苯甲酸为起始原料进行化学合成制备,同时由于硝基的存在,使得反应工艺条件相对温和。从结构来看,目标化合物1是一个四取代苯基结构,由于四个基团紧邻,并且基团本身具有较高的化学活性,所以合成该化合物有一定的难度。关于2,4-二氟-3-硝基苯甲酸的合成,目前未见文献报道,甚至类似结构的合成文献也不多。在专利wo2019053426a1中,公开了一种制备3-乙酰氨基-2,4-二氟苯甲酸的制备方法,该制备方法的反应式如下:经过调研,该路线目前市场上没有起始原料3-氨基-2,4-二氟甲苯的供应,也没有其合成的文献报道,合成难度和成本较高。文献chemistryopen2019,8,166–172中,提供了一种制备间硝基苯甲酸酯方法,该制备方法的反应式如下:该制备方法需要使用特殊制备的催化剂,并且必须以碘代物为起始原料,存在成本高,选择性低和收率低等问题。因此,开发一条原料来源广、成本低、产率高、操作简便以及有利于实现工业化的2,4-二氟-3-硝基苯甲酸的制备方法将具有极强的实用价值。技术实现要素:本发明的目的在于针对上述现有技术的不足,提供一种2,4-二氟-3-硝基苯甲酸合成方法。本发明提供了新颖的式1化合物:本发明的技术方案是:一种2,4-二氟-3-硝基苯甲酸的制备方法,以来源广泛价格低廉的2,6-二氟苯胺(化合物6)为起始原料,先经过氨基保护后得到化合物5;然后化合物5与卤代试剂在适当的溶剂中反应得到化合物4;化合物4在钯催化剂存在下与一氧化碳在温和的条件下进行反应得到化合物3或者化合物3’;化合物3或者化合物3’经过碱性或者酸性水解脱去氨基上的保护基团,同时将生成的羧酸酯或者酰胺水解得到3-氨基-2,4-二氟苯甲酸(化合物2);最后化合物2经过氧化得到2,4-二氟-3-硝基苯甲酸(化合物1),反应式如下:该方法包括下列步骤:步骤1:化合物5的制备氨基保护:其中,r1代表c1-c6的烷基酰基、烷氧羰基、芳烃酰基或取代芳酰基。步骤1:化合物5的制备:在氮气保护下,向反应瓶内加入2,6-二氟苯胺和溶剂,搅拌下控制温度0-100℃滴加氨基保护试剂,滴加结束继续保持反应;反应结束后,经过脱溶结晶得到化合物5;本反应采用的溶剂为甲苯、水、乙酸、二氯甲烷、烷烃,优选甲苯。所述的反应温度为0-100℃,优选0-20℃。所述的反应所使用的氨基保护试剂为c1-c6的氯甲酸酯、c1-c6的烷基酰氯、c1-c6的烷基酸酐和取代或者未取代芳烃酰氯,优选乙酰氯和乙酸酐。步骤2:化合物4的制备卤代部分:其中,x代表氯、溴和碘;r1代表c1-c6的烷基酰基、烷氧羰基、芳烃酰基或取代芳酰基。化合物4的制备:向反应瓶内加入化合物5和溶剂,搅拌至体系全部溶解;分次加入卤代试剂,升温至20-40℃,保持搅拌反应4小时,反应结束,将反应液缓慢倒入冰水中,搅拌析出固体。过滤,固体减压干燥得到化合物4;本反应采用的溶剂为硫酸、乙酸、三氟乙酸,优选硫酸。所述的反应温度为0-100℃,优选20-40℃。所述的反应所使用的卤代试剂为溴素、n-溴代丁二酰亚胺(nbs)、n-碘代丁二酰亚胺(nis)、n-氯代丁二酰亚胺(ncs)和二溴海因(dbdmh),优选nbs。步骤3:化合物3或者化合物3’的制备。其中,x代表氯、溴和碘;r1代表c1-c6的烷基酰基、取代芳烃酰基等。r2代表氢原子、c1-c6的烷基、苯基、取代的苯基。r3和r4代表氢原子、c1-c3的烷基、苯基。化合物3或者化合物3’的制备:将化合物4投入高压釜内,一次性加入碱、溶剂、钯催化剂和膦配体,盖好釜盖,抽真空置换三次;通入一氧化碳气体,升温至50-80℃保持反应8小时。反应结束,将物料降温至室温,用氮气将一氧化碳置换后,过滤将不溶物去除,滤液减压脱溶得到化合物3或者化合物3’;所述的反应温度为20-200℃,优选50-80℃。所述的反应所使用的碱为碳酸钠、碳酸氢钠、碳酸氢钾、三甲胺、三乙胺、三丙胺、三丁胺、二异丙基乙胺、氨气、一甲胺和二甲胺,优选三乙胺。所述的反应所使用的溶剂为甲醇、乙醇、丙醇、异丙醇、丁醇、戊醇、苯酚或者水和甲苯、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、乙腈等,优选为甲醇或水和甲苯,最优选为甲醇。所述的反应所使用的钯为醋酸钯、钯碳或者氯化钯,优选醋酸钯。所述的反应所使用的膦配体为1,1’-双(二苯基膦基)二茂铁、三苯基膦、1,3-双(二苯基膦)丙烷、1,3-双(二苯基膦)丁烷和4,5-双二苯基膦-9,9-二甲基氧杂蒽,优选1,1’-双(二苯基膦基)二茂铁。步骤4:化合物2的制备其中,r1代表c1-c6的烷基酰基、取代芳烃酰基等。r2代表氢原子、c1-c6的烷基、苯基、取代的苯基。r3和r4代表氢原子、c1-c3的烷基、苯基。化合物2的制备:向反应瓶内加入化合物3,溶剂和酸或者碱,升温至80-100℃反应8小时,反应结束,降温至室温,调节ph值为8-9,加入溶剂萃取,有机层废弃,收集水层。水层调节至酸性,加入溶剂萃取,有机层减压脱溶得到化合物2;所述的反应温度为20-200℃,优选80-100℃。所述的水解反应所使用的碱为金属碱,如碳酸钠、氢氧化钠、氢氧化钾、碳酸钾、氢氧化锂,优选为氢氧化钠。所述的水解反应所使用的酸为硫酸、盐酸、三氟乙酸、三氟甲磺酸和乙酸,优选硫酸。步骤5:化合物1的制备化合物1的制备:向反应瓶内加入一定比例的氧化剂,控制温度不超过20℃,滴加3-氨基-2,4-二氟苯甲酸;滴加结束,升温反应至合格后,将反应液缓慢倒入冰水中,加入溶剂萃取,收集有机层,有机层减压脱溶至干,得到2,4-二氟-3-硝基苯甲酸(化合物1)。所述的反应温度为20-100℃,优选20-40℃。所述的反应所使用的氧化剂为双氧水、叔丁基过氧化氢、过氧乙酸、过氧三氟乙酸,优选双氧水。上述中,化合物1为2,4-二氟-3-硝基苯甲酸;化合物2为3-氨基-2,4-二氟苯甲酸;化合物3为3-酰基保护的氨基-2,4-二氟苯甲酸或苯甲酸酯;化合物3’为3-酰基保护的氨基-2,4-二氟苯甲酰胺;化合物4为3-酰基保护的氨基-2,4-二氟卤代苯(x代表氯、溴和碘);化合物5为酰基保护的2,6-二氟苯胺;化合物6为2,6-二氟苯胺。一种2,4-二氟-3-硝基苯甲酸的制备方法,包括以下步骤:首先,化合物六经过氨基保护后得到化合物五;然后,化合物五与卤代试剂在溶剂中反应得到化合物四;其次,化合物四在钯催化剂下与一氧化碳进行反应得到化合物三或者化合物三’;再次,化合物三或者化合物三’经过水解脱去氨基上的保护基团,同时将生成的羧酸酯或者酰胺水解得到化合物二;最后,化合物二经过氧化得到化合物一;其中,化合物一为2,4-二氟-3-硝基苯甲酸;其中,化合物二为3-氨基-2,4-二氟苯甲酸;其中,化合物三为3-酰基保护的氨基-2,4-二氟苯甲酸或苯甲酸酯;其中,化合物三’为3-酰基保护的氨基-2,4-二氟苯甲酰胺;其中,化合物四为3-酰基保护的氨基-2,4-二氟卤代苯(x代表氯、溴和碘);其中,化合物五为酰基保护的2,6-二氟苯胺;其中,化合物六为2,6-二氟苯胺。一种2,4-二氟-3-硝基苯甲酸的制备方法,包括以下步骤:步骤1:以化合物六为原料,在溶剂中进行氨基保护反应得到化合物五;步骤2:化合物五在酸性溶剂中与卤代试剂进行卤代反应得到化合物四;步骤3:化合物四在钯催化剂和膦配体催化下用一氧化碳进行插羰基反应,再与醇、水或者胺进行酯化、水解或者酰胺化反应得到化合物三或者化合物三’(碱性环境下);步骤4:化合物三或者化合物三’,进行水解反应得到化合物二;步骤5:化合物二在酸性溶剂中进行氧化反应得到化合物一。其中,化合物一为2,4-二氟-3-硝基苯甲酸;其中,化合物二为3-氨基-2,4-二氟苯甲酸;其中,化合物三为3-酰基保护的氨基-2,4-二氟苯甲酸或苯甲酸酯;其中,化合物三’为3-酰基保护的氨基-2,4-二氟苯甲酰胺;其中,化合物四为3-酰基保护的氨基-2,4-二氟卤代苯(x代表氯、溴和碘);其中,化合物五为酰基保护的2,6-二氟苯胺;其中,化合物六为2,6-二氟苯胺。进一步,所述步骤1用的溶剂为甲苯、水、乙酸、二氯甲烷和烷烃中的任意一种。进一步,所述步骤1用的反应温度为0-100℃,所述步骤2用的反应温度为0-100℃。进一步,步骤1中使用氨基保护试剂,来实现氨基保护;所述的氨基保护试剂为c1-c6的氯甲酸酯、c1-c6的烷基酰氯、c1-c6的烷基酸酐和取代或者未取代芳烃酰氯中的任意一种。进一步,所述步骤2用的酸性溶剂为硫酸、乙酸、三氟乙酸中的任意一种。进一步,所述步骤2用的卤代试剂为溴素、n-溴代丁二酰亚胺(nbs)、n-碘代丁二酰亚胺(nis)、n-氯代丁二酰亚胺(ncs)和二溴海因(dbdmh)中的任意一种。进一步,所述步骤3用的反应温度为20-200℃。进一步,所述步骤3用的碱为碳酸钠、碳酸氢钠、碳酸氢钾、三甲胺、三乙胺、三丙胺、三丁胺、二异丙基乙胺、氨气、一甲胺和二甲胺中的任意一种。进一步,所述步骤3用的溶剂为甲醇、乙醇、丙醇、异丙醇、丁醇、戊醇、苯酚或者水和甲苯、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、乙腈。进一步,所述步骤3用的钯催化剂为醋酸钯、钯碳或者氯化钯;所述步骤3用的膦配体为1,1'-双(二苯基膦基)二茂铁、三苯基膦、1,3-双(二苯基膦)丙烷、1,3-双(二苯基膦)丁烷和4,5-双二苯基膦-9,9-二甲基氧杂蒽中的任意一种。进一步,所述步骤4用的反应温度为20-200℃。进一步,所述步骤4水解反应为碱性环境,其中,碱性环境所采用的碱为金属碱,如碳酸钠、氢氧化钠、氢氧化钾、碳酸钾、氢氧化锂中的任意一种;或者,所述步骤4水解反应为酸性环境,其中,酸性环境中所采用的酸为硫酸、盐酸、三氟乙酸、三氟甲磺酸、乙酸中的任意一种。进一步,所述步骤5用的反应温度为20-100℃。进一步,步骤5中的氧化反应是通过加入氧化剂来实现;所述步骤5用的氧化剂为双氧水、叔丁基过氧化氢、过氧乙酸、过氧三氟乙酸中的任意一种。本发明的有益效果:第一、本发明涉及一种新的简单的制备3-硝基-2,4-二氟苯甲酸的合成方法,具有原料来源广、成本低、产率高、操作简便,能实现规模工业化的优点。第二、该合成路线未见文献报道,在合成过程中避免使用有毒有害物质,为新药设计和筛选提供价廉的高品质的2,4-二氟-3-硝基苯甲酸合成中间体。附图说明下面结合附图中的实施例对本发明作进一步的详细说明,但并不构成对本发明的任何限制。图1是实施例1的化合物1结构确认的1h-nmr谱图。图2是实施例1的化合物1结构确认的13c-nmr谱图。图3是实施例1的化合物1纯度检测的hplc谱图。具体实施方式通过下述实施例子将有助于本领域技术人员理解本发明的制备技术要点,但是不能限制本发明的内容。实施例1步骤1:2,6-二氟乙酰氨基苯(5)的合成向反应瓶内加入100g(0.77mol)2,6-二氟苯胺和600ml甲苯,搅拌下控制温度0-20℃滴加87g(0.85mol)醋酸酐,滴加结束保持反应4小时,降温至0-10℃,保持搅拌1小时,过滤,固体减压干燥得到2,6-二氟乙酰氨基苯(5)125g,收率为94%。步骤2:3-溴-2,6-二氟乙酰氨基苯(4)的合成向反应瓶内加入55g(0.32mol)2,6-二氟乙酰氨基苯和660g浓硫酸,搅拌至体系全部溶解。20-40℃下,分次加入63g(0.35mol)nbs,加完保持搅拌反应5h。待反应合格后,将反应液缓慢倒入1000g冰水中,搅拌析出白色固体。过滤得到3-溴-2,6-二氟乙酰氨基苯粗品。向3-溴-2,6-二氟乙酰氨基苯粗品中加入220ml醋酸异丙酯,升温至回流,保持搅拌2小时,再降温至0-10℃,保持搅拌1小时。过滤,减压干燥得到3-溴-2,6-二氟乙酰氨基苯(4)69g,收率为86%。步骤3:3-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的合成将20g(0.08mol)3-溴-2,6-二氟乙酰氨基苯投入高压釜内,一次性加入57g三乙胺、200ml甲醇、0.1g醋酸钯和0.4g1,1'-双(二苯基膦基)二茂铁。盖好釜盖,抽真空置换三次;通入一氧化碳气体,升温至50-80℃,保持反应8h。反应结束,将物料降温至室温,用氮气将一氧化碳置换后,过滤,滤液减压脱溶得到3-乙酰氨基-2,4-二氟苯甲酸甲酯(3)16.7g,收率为93%。步骤4:3-氨基-2,4-二氟苯甲酸(2)的合成向反应瓶内加入32g(0.14mol)3-乙酰氨基-2,4-二氟苯甲酸甲酯,260g20%硫酸和60ml甲苯,升温至80-100℃,搅拌反应4小时。待反应结束后,控制温度不超过30℃,滴加氢氧化钠溶液,调节ph值为8-9。加入100ml甲苯萃取。水层滴加硫酸,调节ph值为3.0-3.5。加入600ml醋酸异丙酯萃取。减压脱溶得到3-氨基-2,4-二氟苯甲酸(2)21.5g,收率为90%。步骤5:2,4-二氟-3-硝基苯甲酸(1)的合成向反应瓶内加入40g30%双氧水,控制温度不超过20℃,滴加10g(0.06mol)3-氨基-2,4-二氟苯甲酸与50g三氟乙酸配制成的溶液,滴加结束,控温20~40℃保持反应5小时。待反应结束后,将反应液缓慢倒入200g冰水中,搅拌升温至20-30℃,加入200g醋酸异丙酯萃取,加入活性炭,搅拌30分钟,过滤,滤液减压脱溶至干,加入5ml醋酸异丙酯和50ml庚烷,升温至40-50℃,搅拌0.5小时,然后降温至0-5℃,搅拌1小时。然后过滤,减压干燥得到2,4-二氟-3-硝基苯甲酸(1)10.3g,收率为88%,纯度为99.7%。1hnmr(dmso-d6),400mhz,δ13.91(s,2h),δ8.23-8.17(m,1h),δ7.56-7.52(t,1h)。其中,化合物1的1h-nmr和13c-nmr和纯度检测的hplc谱图,见附图1、2、3。实施例22,6-二氟乙酰氨基苯(5)的合成按照实施例1的步骤1进行操作,只是反应溶剂分别为甲苯、乙酸、二氯甲烷和己烷,其他操作条件一致。2,6-二氟乙酰氨基苯(5)的收率与不同溶剂的数据,见表1。表1反应溶剂反应时间(h)反应收率%甲苯494乙酸480二氯甲烷490己烷860实施例32,6-二氟乙酰氨基苯(5)的合成。按照实施例1的步骤1进行操作,只是反应温度分别为0-20℃、20-40℃和60-80℃,其他操作条件一致。2,6-二氟乙酰氨基苯(5)的收率与不同温度的数据,见表2。表2反应温度(℃)反应时间(h)反应收率%0-2049420-4049260-80488实施例4酰基保护的2,6-二氟苯胺(5)的合成。按照实施例1的步骤1进行操作,只是氨基保护基分别为醋酸酐、乙酰氯、氯甲酸甲酯和苯甲酰氯,其他操作条件一致。酰基保护的2,6-二氟苯胺(5)的收率与不同氨基保护基的数据,见表3。表3氨基保护基反应时间(h)反应收率%醋酸酐494乙酰氯493氯甲酸甲酯488苯甲酰氯492实施例53-溴-2,6-二氟乙酰氨基苯(4)的合成。按照实施例1的步骤2进行操作,只是卤代试剂分别为n-溴代丁二酰亚胺、溴素和二溴海因,其他操作条件一致。3-溴-2,6-二氟乙酰氨基苯(4)的收率与卤代试剂的数据,见表4。表4卤代试剂反应时间(h)反应收率%n-溴代丁二酰亚胺586溴素580二溴海因576实施例63-溴-2,6-二氟乙酰氨基苯(4)的合成。按照实施例1的步骤2进行操作,只是反应溶剂分别为浓硫酸、乙酸和三氟乙酸,其他操作条件一致。3-溴-2,6-二氟乙酰氨基苯(4)的收率与不同反应溶剂的数据,见表5。表5反应溶剂反应时间(h)反应收率%浓硫酸586乙酸560三氟乙酸565实施例73-溴-2,6-二氟乙酰氨基苯(4)的合成。按照实施例1的步骤2进行操作,只是反应温度分别为0-20℃、20-40℃和60-80℃,其他操作条件一致。3-溴-2,6-二氟乙酰氨基苯(4)的收率与不同反应温度的数据,见表6。表6反应温度反应时间(h)反应收率%0-2057520-4058660-80555实施例83-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的合成。按照实施例1的步骤3进行操作,只是催化剂分别为醋酸钯、5%钯碳和氯化钯,其他操作条件一致。3-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的收率与不同催化剂的数据,见表7。表7催化剂反应时间(h)反应收率%醋酸钯8935%钯碳860氯化钯888实施例93-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的合成。按照实施例1的步骤3进行操作,只是膦配体分别为1,1’-双(二苯基膦基)二茂铁、三苯基膦和4,5-双二苯基膦-9,9-二甲基氧杂蒽,其他操作条件一致。3-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的收率与不同膦配体的数据,见表8。表8膦配体反应时间(h)反应收率%1,1’-双(二苯基膦基)二茂铁893三苯基膦8804,5-双二苯基膦-9,9-二甲基氧杂蒽890实施例103-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的合成。按照实施例1的步骤3进行操作,只是反应使用的碱分别为三乙胺、二异丙基乙胺和碳酸钠,其他操作条件一致。3-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的收率与不同碱的数据,见表9。表9反应使用的碱反应时间(h)反应收率%三乙胺893二异丙基乙胺892碳酸钠885实施例11。3-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的合成。按照实施例1的步骤3进行操作,只是反应温度分别为20-40℃、50-80℃、80-100℃和140-160℃,其他操作条件一致。3-乙酰氨基-2,4-二氟苯甲酸甲酯(3)的收率与不同反应温度的数据,见表10。表10反应温度反应时间(h)反应收率%20-4086050-8089380-100892140-160879实施例123-乙酰氨基-2,4-二氟苯甲酰胺(3’)的合成。将20g(0.08mol)3-溴-2,6-二氟乙酰氨基苯投入高压釜内,一次性通入50g氨气、200ml甲苯、0.1g醋酸钯和0.4g1,1'-双(二苯基膦基)二茂铁。盖好釜盖,氮气置换三次;通入一氧化碳气体,升温至50-80℃,保持反应8h。反应结束,将物料降温至室温,用氮气将一氧化碳置换后,过滤,滤液减压脱溶得到3-乙酰氨基-2,4-二氟苯甲酰胺15.4g,产率为90%。实施例133-乙酰氨基-2,4-二氟苯甲酸的合成。将20g(0.08mol)3-溴-2,6-二氟乙酰氨基苯投入高压釜内,一次性加入57g三乙胺、100ml水、100甲苯、0.1g醋酸钯和0.4g1,1'-双(二苯基膦基)二茂铁。盖好釜盖,抽真空置换三次;通入一氧化碳气体,升温至50-80℃,保持反应8h。反应结束,将物料降温至室温,用氮气将一氧化碳置换后,过滤,滤液减压脱溶得到产品15.5g,产率为90%。实施例143-氨基-2,4-二氟苯甲酸(2)的合成。按照实施例1的步骤4进行操作,只是反应温度分别为20-40℃、40-60℃和80-100℃,其他操作条件一致。3-氨基-2,4-二氟苯甲酸(2)的收率与不同反应温度的数据,见表11。表11反应温度反应时间(h)反应收率%20-40206040-60157080-100490实施例153-氨基-2,4-二氟苯甲酸(2)的合成。表12水解反应试剂反应时间(h)反应收率%硫酸490盐酸780三氟甲磺酸487乙酸1540氢氧化钠530碳酸钾550实施例15的条件:按照实施例1的步骤4进行操作,只是水解反应使用的试剂分别为硫酸、盐酸、三氟甲磺酸、乙酸、氢氧化钠和碳酸钾,其他操作条件一致。3-氨基-2,4-二氟苯甲酸(2)的收率与不同水解试剂的数据,见表12。实施例162,4-二氟-3-硝基苯甲酸(1)的合成。按照实施例1的步骤5进行操作,只是氧化反应温度分别为0-20℃、20-40℃、40-60℃和60-80℃,其他操作条件一致。2,4-二氟-3-硝基苯甲酸(1)的收率与不同温度的数据,见表13。表13反应温度(℃)反应时间(h)反应收率%0-20106520-4058840-6058260-80460从表13可知,反应温度对于反应收率的影响较大,在反应温度在0-20°时,反应时间10h后的反应收率仍然只有65%;而反应温度在20-40°时,反应时间10h后的反应收率有88%。从表13可知,反应温度对于反应收率的影响,是先增大,后减小;即在温度大于一定值后,其对反应又不利。实施例172,4-二氟-3-硝基苯甲酸(1)的合成。按照实施例1的步骤5进行操作,只是氧化剂分别为双氧水、叔丁基过氧化氢、过氧乙酸和过氧三氟乙酸,其他操作条件一致;2,4-二氟-3-硝基苯甲酸(1)的收率与不同氧化剂的数据,见表14。表14氧化剂反应时间(h)反应收率%双氧水588叔丁基过氧化氢580过氧乙酸540过氧三氟乙酸550从表14可知:采用双氧水的反应收率最高,能够达到88%;叔丁基过氧化氢次之,达到80%;而过氧三氟乙酸、过氧乙酸相对较差,分别只有50%、40%。需要说明的是,本发明的产品纯度检测(实施例1生产的产品)采用如表所示的hplc条件,见表15。表15以上所举实施例为本发明的较佳实施方式,仅用来方便说明本发明,并非对本发明作任何形式上的限制,任何所属
技术领域:
中具有通常知识者,若在不脱离本发明所提技术特征的范围内,利用本发明所揭示技术内容所作出局部更动或修饰的等效实施例,并且未脱离本发明的技术特征内容,均仍属于本发明技术特征的范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1