干扰柑橘木虱卵黄蛋白原基因表达的转基因生防真菌及其制备方法和应用与流程
本发明涉及基因工程及害虫防治技术领域,具体涉及一种干扰柑橘木虱卵黄蛋白原基因表达的转基因生防真菌及其制备方法和应用。
背景技术:
柑橘黄龙病是毁灭性柑橘病害,柑橘木虱(diaphorinacitrikuwayama)是柑橘黄龙病最为重要的传播媒介,防治柑橘木虱是控制柑橘黄龙病的关键措施。目前,化学防治仍然是柑橘木虱防控中的主要手段,但化学防治极易造成农药残留、环境污染、害虫抗药性等问题。生物防治是克服化学防治缺点的重要补充途径,微生物(丝状真菌)防治是生物防治的一个重要途径,它主要是利用昆虫病原真菌对害虫进行侵染,穿透其体壁并在其体内萌发生长,最终导致其死亡的一种防治方式。
渐狭蜡蚧菌lecanicilliumattenuatum是一种重要的生防真菌,对柑橘木虱具有控制作用,渐狭蜡蚧菌可以寄生蚜虫、粉虱、木虱、线虫等害虫(螨)以及一些植物病原菌,可是目前对该菌的研究较少。渐狭蜡蚧菌天然菌株浸染害虫速度慢,实际防治效果不太理想,因此,需要对渐狭蜡蚧菌进行遗传改良,提高其对柑橘木虱的毒力。
卵黄蛋白原(vitellogenin,vg)几乎是所有卵生动物卵黄蛋白(vitellin,vn)的前体,它普遍存在于卵生非哺乳类性成熟动物的血液中,参与卵生动物的发育和生殖等过程。rna干扰(rnainterfere,rnai)是由同源的dsrna介导的转录后基因沉默现象。在昆虫rnai的研究应用中,直接注射dsrna、饲喂dsrna和构建rnai载体是目前使用rnai技术的主要方法。
目前对于利用真菌作为媒介干扰害虫(螨)的研究很少,对这些昆虫的干扰研究主要集中在含有昆虫靶基因dsrna的转基因植物中。在很多转基因的植物中都取得了成功,起到了很好防治昆虫的作用,mao等人就通过转棉铃虫p450基因到植物中,使得对该虫有了一个良好的控制效应(maoetal.,2007)。但是也有转入的基因不执行rnai功能,张凤珍就将褐飞虱的基因转入水稻中,但是并没有发现对褐飞虱有rnai效应(张凤珍,2013)。在实验中,对于rani不执行其功能的原因很多,不确定因素也很多,有的研究人员认为可能是dsrna释放效果不佳或rnai载体中的intron匹配度低等原因导致的,目前关于此的报道并不是很多,其原因需要更多的科学研究去发现。
技术实现要素:
本发明的目的是针对上述问题,利用现代生物技术构建柑橘木虱卵黄蛋白原基因的rnai载体,并将其导入生物防治真菌基因组中,从而缩短生防真菌对柑橘木虱的浸染时间,提高生防真菌对柑橘木虱的毒力,方案具体如下:
一种柑橘木虱卵黄蛋白原基因rnai载体,包括骨架载体和发夹结构,所述发夹结构为ihprna表达盒,所述ihprna表达盒的结构为ptrpc启动子+正向目的基因片段+intron+反向目的基因片段+ttrpc终止子结构,所述目的基因片段为柑橘木虱卵黄蛋白原基因保守序列dcvg1a或dcvg1b;所述骨架载体为丝状真菌表达载体。
所述丝状真菌表达载体是利用pdmⅰ或者xmnⅰ、和pstⅰ2种酶对prcy1载体进行酶切,获得含有hygr、intron片段的线性载体,再利用重组位点将ptrpc启动子+egfp片段连入prcy1线性载体中得到的。
上述的柑橘木虱卵黄蛋白原基因rnai载体的构建方法:采用一步克隆法将ptrpc启动子+egfp基因片段连接到丝状真菌表达载体prcy1中获得prcy3,用一步克隆法将目的基因的正向片段转入prcy3载体启动子和intron之间,然后用一步克隆法转入反向片段,即得。
在上述技术方案中,所述ptrpc启动子+egfp基因片段是以prcy2a为模板,设计含有xmnⅰ和pstⅰ酶酶切位点的引物,进行pcr扩增得到;
利用pdmⅰ或者xmnⅰ、和pstⅰ2种酶对prcy1载体进行酶切获得线性载体,利用重组位点用一步克隆法将得到的ptrpc启动子+egfp片段连入prcy1线性载体中,构建成含有egfp、intron结构的丝状真菌双元载体prcy3;
先将prcy3载体进行xhoⅰ和smiⅰ双酶切,然后将目的基因的正向片段连入双酶切后的prcy3,然后将连接有正向片段的载体用xbai和bamhi进行酶切后连入对应的反向目的片段。
本发明的再一目的在于提供上述的rnai载体在防治柑橘木虱中的应用、在制备防治柑橘木虱的产品中的应用、在制备转基因生防真菌中的应用;
优选所述生防真菌为渐狭蜡蚧菌。
本发明的再一目的在于提供一种转基因生防真菌:是采用前述任一的rnai载体转化生防真菌得到。
优选地,所述生防真菌为渐狭蜡蚧菌,所述转化是由农杆菌介导;
进一步优选地,所述农杆菌为agl1。
本发明的再一目的在于提供上述的转基因渐狭蜡蚧菌的制备方法:采用上述任一的rnai载体转化农杆菌,随后利用诱导培养法将农杆菌诱导培养后与生防真菌孢子悬浮液共培养,分离、检测验证,得到转基因成功的生防真菌菌株。
本发明的再一目的在于提供上述的转基因生防真菌在防治柑橘木虱中的应用、在制备防治柑橘木虱的产品中的应用。
本发明的再一目的在于提供一种柑橘木虱防治制剂:包含上述任一的rnai载体,或者上述任一的的转基因生防真菌。
本发明的有益效果是:选用柑橘木虱卵黄蛋白原基因保守序列dcvg1a或dcvg1b构建发夹结构,选用丝状真菌表达载体作为骨架载体,通过特定的限制性内切酶酶切骨架载体,成功构建了柑橘木虱卵黄蛋白原基因rnai载体,使得最终获得的rnai载体具有特定的ihprna表达盒结构(ptrpc启动子+正向目的基因片段+intron+反向目的基因片段+ttrpc终止子结构),利用该rnai载体转化生防真菌渐狭蜡蚧菌,得到的转基因渐狭蜡蚧菌经实验证实可有效提高其对柑橘木虱毒力的毒力,缩短对木虱的浸染时间。
附图说明
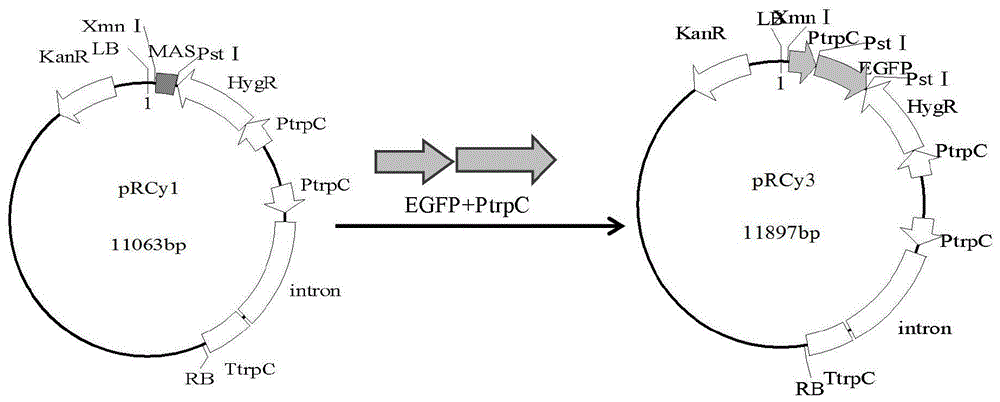
图1是ptrpc启动子+egfp替换prcy1载体中的mas的载体构架框架图。
图2是卵黄蛋白原基因rnai载体构建图。
图3是柑橘木虱vg基因的保守序列分析。
图4是agl1::gfp菌株的菌液pcr验证结果,其中,泳道1、2、3分别为三个agl1::gfp阳性菌株检测到的潮霉素抗性基因阳性条带。
图5是agl1::dcvg1a和agl1::dcvg1b菌株的菌液pcr和酶切验证结果,其中,图a为菌液pcr结果,1-6泳道为agl1::dcvg1a、7-12泳道为agl1::dcvg1b;图b为酶切验证结果,泳道1、2为agl1::dcvg1a菌株质粒提取后酶切下来的目的片段;泳道3、4为agl1::dcvg1b菌株质粒提取后酶切下来的目的片段。
图6是农杆菌介导的渐狭蜡蚧菌转化过程示意图。
图7是转基因菌株的菌丝快速鉴定结果,其中,图a是egfp检测引物结果,泳道1-3为渐狭蜡蚧菌la::dcvg1a阳性菌株所含egfp基因条带;泳道4-5为渐狭蜡蚧菌la::dcvg1b阳性菌株所含egfp基因条带;图b是潮霉素检测引物结果,泳道1-5为渐狭蜡蚧菌la::dcvg1a阳性菌株所含抗潮霉素基因条带;泳道6-12为渐狭蜡蚧菌la::dcvg1b阳性菌株所含抗潮霉素基因条带。
具体实施方式
下面结合实施例对本发明作进一步说明,但并不因此而限制本发明。
下述实施例中的实验方法,如无特别说明,均为常规方法。
主要试剂来源:
prcy1质粒:本实验室保存,其构建方法参照硕士论文“于士将.渐狭蜡蚧菌(lecanicilliumattenuatum)介导的rnai对柑橘粉虱毒力探究[d].西南大学,2015.”;
prcy2a质粒:本实验室保存,其构建方法参照硕士论文“杨涓.柑橘粉虱卵黄蛋白原基因rnai载体构建及对渐狭蜡蚧菌(lecanicilliumattenuatum)的转化[d].西南大学,2016.”;
2×phantamaxmastermix:南京诺唯赞生物科技股份有限公司,货号p515-01;
clonexpressⅱonestepcloningkit试剂盒:南京诺唯赞生物科技股份有限公司,货号c112-01;
内切酶pdmⅰ、xmnⅰ、pstⅰ、xhoⅰ、smiⅰ、bamhⅰ、xbaⅰ:thermofisherscientific公司产品;
dna胶纯化回收试剂盒:美国omega公司产品;
totalrnakit试剂盒:美国omega公司产品;
agl1农杆菌感受态细胞:北京博迈德基因技术有限公司;
渐狭蜡蚧菌菌株tn002:本实验室保存。
其余试剂如未表明,均为本领域常规试剂,可商购获得。
实施例1丝状真菌表达载体的构建与改造
为了在转基因菌株评价中可以更加直观的观察到载体在真菌中的表达情况,根据同源重组原理选择一步克隆法将ptrpc启动子+egfp基因片段连接到实验室原有的丝状真菌表达载体prcy1中,获得有报告基因egfp(增强型绿色荧光蛋白)的丝状真菌表达载体,命名为prcy3。对柑橘木虱转录组数据库进行筛选,发现柑橘木虱卵黄蛋白原基因的保守序列dcvg1和dcvg2,根据同源重组原理设计引物,用一步克隆法将目的基因的正向片段转入prcy3载体启动子和intron之间,用正向检测引物进行菌液pcr、酶切验证和测序验证。用相同的方法转入反向片段。验证成功后获得有ptrpc启动子+正向目的片段+intron+反向目的片段+ttrpc终止子结构的丝状真菌表达载体,完成ihprna表达盒的构建。
1.含egfp基因的载体改造
1.1载体框架
载体框架如图1所示。
以prcy2a为模板,设计含有xmnⅰ和pstⅰ酶酶切位点的引物,用2×phantamaxmastermix扩增出含ptrpc+egfp结构片段备用,利用pdmⅰ(xmnⅰ的同工酶)和pstⅰ2种酶对prcy1载体进行酶切,获得含有hygr(潮霉素抗性)、intron片段的线性载体,利用重组位点将ptrpc启动子+egfp片段连入prcy1线性载体中,构建成含有egfp、intron结构的植物双元载体prcy3。
1.2含重组位点的ptrpc启动子+egfp(长度1098bp)的克隆
根据ptrpc启动子+egfp序列和prcy2a载体序列,利用引物设计软件primer5.0设计含prcy1上2个酶切位点两端15bp的重组位点的ptrpc启动子+egfp序列的扩增引物f:5’-tactgaattaacgccgaattaattcggatgcttgggtagaataggtaa-3’(seqidno:3),r:5’-atagagtagatgccgctgcagatggtgagcaagggcgagg-3’(seqidno:4)。对目的片段进行扩增,扩增体系为:2×phantamaxmastermix25μl,引物f2μl,引物r2μl,模板prcy2a2μl,ddh2o至19μl。混匀后在pcr仪上进行扩增,94℃预变性3min;94℃变性30s,68.3℃退火30s,72℃延伸30s,循环40次;72℃彻底延伸5min后放4℃保存备用。
制作1%的琼脂凝胶45ml,将pcr产物在凝胶中上样,用bio-red电泳仪检测(110v,30min),在紫外成像仪下观察结果,然后进行切胶回收,用dna胶纯化回收试剂盒进行产物回收,得到ptrpc+egfp片段。
1.3prcy1线性载体的获得
用实验室保存的含prcy1的质粒,测序验证prcy1中载体位点序列。用snapgene软件对prcy1载体序列分析比对后,找出1对特异且唯一的酶pdmⅰ和pstⅰ,对prcy1载体进行酶切。反应程序为:prcy1质粒5μg、10×buffer5μl、pdmⅰ2μl、pstⅰ2μl、ddh2o加至50μl,混匀后在37℃恒温金属浴中酶切2h后立即置于冰上。取2μl酶切反应液电泳检测酶切完整度,确定完全酶切后利用柱回收试剂盒回收载体线性片段,步骤如下:
(1)在酶切反应液中加入3-5倍体积的xp5buffer,低速离心混匀;
(2)将混液上柱于hibinddnacolumn,10000xg常温离心1min,弃滤液;
(3)加入700μlwashbufferspw漂洗柱子,10000xg离心1min,弃滤液;重复洗脱一次;
(4)加入30μlelutionbuffer,13000xg离心2min;
(5)用thermonandrop2000分光光度计测量回收产物浓度,放入-20冰箱保存备用。
1.4同源重组连接
用clonexpressⅱonestepcloningkit试剂盒将ptrpc+egfp片段和prcy1线性载体同源重组连接。反应体系为线性化载体50-200ng、插入片段20-200ng、5×cebuffer2μl、exnaseⅱ1μl、ddh2o加至10μl。将反应液至于37℃金属浴孵育30min后立即置于冰上冷却5min,将大肠杆菌感受态细胞从-80℃超低温冰箱取出冻融,吸取100μl感受态加入到反应液中,轻轻混匀,冰上静置30min。将混液在42℃恒温热水浴中热激45s,立即取出,冰上冷却2-3min,室温静置3min。为使载体上的抗性复苏,向反应液中加入900μl不含抗生素的lb液体,于37℃,200rpm/min摇床孵育1h。取出后5000xg室温离心5min,吸除900μl上清液,将剩余反应液吹打混匀后涂布于含kan的lb固体培养基上,倒置暗培养12h后挑取阳性单菌落,用egfp检测引物(序列如表2中edp所示引物)进行菌液pcr验证,后提取质粒进行酶切验证,验证正确的质粒命名为prcy3。
2.柑橘木虱卵黄蛋白原基因片段克隆
2.1载体框架
卵黄蛋白原基因rnai载体构建图如图2所示。
2.2柑橘卵黄蛋白原基因的查找
通过筛查实验室柑橘木虱转录组数据库中有关卵黄蛋白原的相关基因,在ncbi数据库中比对后筛选出柑橘木虱卵黄蛋白原vitellogenin-a1-like基因(genebank登录号:xm_026832896)。柑橘木虱vg-a-likecdna序列全长4808bp,开放阅读框长度4602bp,编码1534个氨基酸残基。ncbi上conserveddomains分析后结果如图3所示,该序列包含3个传统的保守域vit-n(84-410aa)、vit-n(603-920aa)、vit-n(1205-1520aa)。分析比较后发现该基因中编码前两个保守域的基因具有高度相似性,因此选择了vit-n(84-410aa)和vit-n(1205-1520aa)作为目的基因,标记为dcvg1a、dcvg1b,其核酸序列分别为seqidno:1和seqidno:2所示。
2.3引物设计
利用prime5.0和同源重组的方法设计2个基因的正反向扩增引物(含prcy3载体上特有的酶切位点两端15bp重组位点),引物序列如表1所示。为检测靶标基因目的片段是否成功转入载体中,在酶切位点之外设计了正反向通用的检测引物,用于检测靶标基因是否正确插入载体中,引物序列如表2所示。
表1柑橘木虱卵黄蛋白原基因的正反向重组引物
表2载体检测引物
2.4对prcy3载体进行xhoⅰ和smiⅰ双酶切
用xhoⅰ和smiⅰ核酸内切酶对prcy3载体进行酶切,具体操作步骤按照试剂说明书进行。取出2μl电泳检测,若获得的目的条带单一无杂带,将剩余反应液用胶纯化回收试剂盒回收,进行测序验证。
2.5柑橘木虱总rna的提取
柑橘木虱总rna的提取,利用totalrnakit试剂盒提取柑橘木虱成虫总rna。步骤如下:
将柑橘木虱成虫从-80℃取出后,在液氮环境下,用rnase-free的研磨棒将其研磨成粉末状;加入350μl2-巯基乙醇裂解液trklysisbuffer,离心混匀,静置5min;待其充分混匀后在4℃条件下13000xg离心3min;将上清液转移到新的1.5mlrnase-free离心管中,加入等体积的70%乙醇(事先低温保存),充分混匀;转移到rna过滤柱中静置2min后进行过柱,10000xg离心30s;加入400μlrwcwashbuffer(使用前确保加入无水乙醇),静置2min,10000xg离心30s后弃滤液;向柱中滤膜上滴加10μldnase去除其中的dna;加入rwbwashbuffer500μl,10000xg离心30s,弃滤液,重复一次;最大转速离心2min,将残余的无水乙醇晾干;用30μldepc水洗脱柱上的rna,取2μl电泳检测其完整性。用thermonandrop分光光度计测量rna浓度,放入-80℃保存备用。
2.6柑橘木虱cdna合成
用反转录试剂盒(南京诺唯赞生物科技股份有限公司产品)将柑橘木虱rna样品反转录合成cdna,步骤如下:
在rnase-free离心管中加入9μlrnase-freeddh2o、4μl4×gdnawipermix、1μloligo(dt)23vn(50μm)、1μlrandomhexamers(50ng/μl)和1μg2.3获得的rna(此过程须在冰上操作)后用移液器轻轻吹打混匀。42℃热孵育2min去除rna中的基因组残留。向上一步混液中加入10×rtmix和hiscriptiienzymemix各2μl。混匀后在pcr仪上进行如下反应:50℃15min,85℃2min。产物放在-40℃冰箱备用。
2.7靶标基因克隆与连接以柑橘木虱cdna为材料,用表1的引物分别扩增所需目的片段(dcvg1a正向/dcvg1a反向、dcvg1b正向/dcvg1b反向),扩增体系为:2×phantamaxmastermix25μl,浓度为10μm的正、反向引物各2μl,柑橘木虱cdna2μl,ddh2o至19μl。
扩增dcvg1a正向/dcvg1a反向、dcvg1b正向/dcvg1b反向片段的pcr反应程序均为:预变性:95℃,3min;变性:95℃15s,退火:60℃15s,延伸:72℃30s,35个循环;彻底延伸:72℃,5min。
以prcy3作为基础载体,用同源重组连接的方法将卵黄蛋白原dcvg1a或者dcvg1b的正向片段连入双酶切(xhoi和smii)后的prcy3,具体方法参照前面“1.4同源重组连接”部分的步骤。验证连接成功后,将含有卵黄蛋白原基因正向片段的prcy3用xbai和bamhi进行酶切后采用相同的方法连入对应的反向目的片段。
用表2中的引物进行验证,用引物fdp验证dcvg1a或者dcvg1b的正向片段,用引物rdp验证dcvg1a或者dcvg1b的反向片段。验证成功后获得有ptrpc启动子+正向目的片段+intron+反向目的片段+ttrpc终止子结构的丝状真菌表达载体(即rnai质粒),即完成ihprna表达盒的构建。同时制备egfp的ihprna表达盒作为对照。
实施例2农杆菌介导的ihprna表达盒转化渐狭蜡蚧菌
利用冻融法,将构建成功的ihprna表达盒进行转化,利用培养基筛选、引物扩增和酶切的方法对农杆菌菌株agl1阳性克隆进行验证,获得含有ihprna表达盒的农杆菌菌株。随后,进一步利用乙酰丁香酮诱导培养法,将农杆菌诱导培养6h后与渐狭蜡蚧菌孢子悬浮液共培养60h左右,将其转移到潮霉素选择性培养基上继续培养,并分离纯化、pcr检测,发现ihprna表达盒已成功整合到渐狭蜡蚧菌基因组中。测序结果表明,该渐狭蜡蚧菌菌株含柑橘木虱卵黄蛋白原基因ihprna表达盒,渐狭蜡蚧菌的遗传改造完成。
一、具体方法步骤:
1.冻融法转化农杆菌agl1
(1)于-80℃超低温冰箱中取出agl1感受态细胞,0℃冰上冻融10min,吸取实施例1已经构建好的rnai质粒1ug,加入到感受态细胞中,吹打混匀,在冰上静置5min;
(2)立即取出在液氮中冷冻5min;
(3)迅速取出放在恒温金属浴上,37℃孵育5min,中途不得摇晃移动;
(4)取出后立即放在冰上静置5min;
(5)在超净工作台中向离心管中加入800μl无任何抗生素的lb液体培养基,吹打混匀;封上封口膜;
(6)将混液置于28℃,200转得恒温摇床中孵育2h;
(7)取出混液,6000xg常温离心5min,吸除800μl上清液,将剩余的液体吹打混匀,涂布在含kan的lb固体培养基上,置于28℃培养箱避光培养;
(8)24h后挑取单菌落于含kan的lb液体培养基中培养,扩大菌群量后通过菌液pcr、提质粒酶切验证等方式筛选阳性转化子。
2.农杆菌介导转化渐狭蜡蚧菌
(1)纯化农杆菌菌株。将含有目的基因片段的agl1菌株重活菌体后,在含kan的lb培养基上划线纯化。24害后挑取单菌落验证。
(2)im诱导培养农杆菌。取100μl菌液扩大培养,28℃200转恒温摇床摇至其od值0.2左右时,6000xg离心5min收集菌体,加入3mlim培养基,吹打混匀后在28℃、200rpm/min恒温摇床中诱导6h。
(3)渐狭蜡蚧菌tn002菌株孢子悬浮液制备。选取重新分离出来的渐狭蜡蚧菌菌株,用0.05%吐温-80配置孢子悬浮液,用血球计数板计数,配置成浓度为1.0×106孢子/ml的悬浮液。
(4)cm固体培养基制备。将配好的cm培养基倒平板,并在中央放置一张灭菌的定量滤纸备用。
(5)共培养。取等体积im诱导混液和渐狭蜡蚧菌孢子悬浮液混匀,取200μl的混液均匀涂布在cm固体培养基滤纸上,放入25℃黑暗培养48h。
(6)选择性培养。将上一步的滤纸取出正放在含有0.40ml0.1g/ml头孢霉素、1ml50mg/ml潮霉素b的200mlpda选择培养基中,28℃培养至滤纸上可见单菌落(约3d)。
(7)纯化。用接种针刮下滤纸上的菌落,转移到新的筛选培养基上,培养4-5d后打菌饼配制成孢子悬浮液,稀释成1×103孢子/ml,吸取100μl涂布到pda选择培养基上进行单孢分离,28℃暗培养。
(8)pcr验证。将单菌落转接到选择培养基上扩大种群,挑取单菌落用潮霉素检测引物进行pcr验证,引物为hygr-f:5’-ctacacagccatcggtccag-3’(seqidno:19),hygr-r:5’-gttcgcccttcctcccttta-3’(seqidno:20)。
(9)酶切验证。用天根公司的质粒小提试剂盒提取各菌株质粒,用pstⅰ进行单酶切验证含prcy3载体菌株,用bamhⅰ和xbaⅰ进行双酶切验证含目的基因载体的菌株。
(10)测序。送擎科生物公司测序验证。
二、结果:
1.含ihprna表达盒的农杆菌制备
用冻融法将载体转入农杆菌agl1后,对农杆菌进行划线培养获取单菌落,对其进行菌液pcr验证和酶切验证,在用潮霉素检测引物在1030bp附近获得了阳性条带(图4)。后提取农杆菌质粒,用pstⅰ核酸内切酶对质粒进行单酶切,由于质粒量太少,电泳条带不是很清晰。后送擎科生物公司测序验证成功后备用。对导入含卵黄蛋白原基因的载体的农杆菌也进行了反向检测引物的菌液pcr验证(图5a,其中泳道2和7为假阳性扩增),发现agl1::dcvg1a菌株中各菌落均能在1125bp处扩增出阳性条带,但发现2和3的条带与目的条带大小不符,故舍弃。agl1::dcvg1b菌株中除第一个菌落(对应条带7)无阳性片段外,其余菌落均能在1095bp处扩增到阳性条带。后各挑取agl1::dcvg1a、agl1::dcvg1b两个菌株中的2个菌落摇菌,提取质粒,用xbaⅰ和bamhⅰ对其进行酶切,结果如图5b所示,在1000bp附近酶切出单一的阳性条带。之后对质粒的插入片段进行测序,序列验证正确。
2.农杆菌介导法介导的渐狭蜡蚧菌转基因菌株
农杆菌介导的渐狭蜡蚧菌转化过程如图6所示,通过农杆菌冻融法将对照载体和3个靶标载体转入agl1菌株成功后,将农杆菌在28℃,200rpm/min摇床中培养至菌液浓度od=0.2左右时,进行诱导培养6h左右(im培养基),取等体积诱导培养液与新鲜的1.00×106孢子/ml的渐狭蜡蚧菌孢子悬浮液混合,均匀涂布在铺有定量滤纸的共培养基上,25℃黑暗培养60h左右。将长出的单菌落真菌转接到含有潮霉素的pda选择性培养基上培养4d,此时如果载体传入成功,菌株基因组中含有潮霉素基因,菌株就能在潮霉素选择性培养基上生长,若载体导入不成功,菌株将无法在选择性培养基上快速生长。至此可以初步筛选出载体导入成功的菌株。
将转接成功的单菌落真菌挑取到新的pda选择性培养基上,待菌落扩大后用egfp检测引物和潮霉素检测引物进行菌丝快速鉴定,结果如图7所示,发现均能扩增出阳性条带。
实施例3转基因菌株的实际效果
在完成对渐狭蜡蚧菌的遗传改造的基础上,对其生物学特性和改造效果进行评价。结果发现,转基因菌株la::dcvg1a和la::dcvg1b的生长速率和单位产孢量,均与野生型菌株无显著性差异,说明柑橘木虱卵黄蛋白原基因的转入对渐狭蜡蚧菌生物学特性无显著影响。在无抗生素的pda培养基上连续培养10代后用潮霉素检测引物扩增,在1030bp处均能扩增出阳性条带,说明农杆菌介导转化法具有很好的稳定性。
用实施例2得到的转基因渐狭蜡蚧菌孢子悬浮液浸没处理柑橘木虱雌成虫15s,随后,每24h收集一次雌成虫,进行rt-qpcr,检测卵黄蛋白原基因的相对表达量,以0.05%吐温-80溶液处理作对照,与la::gfp菌株相比,转基因菌株la::dcvg1a和la::dcvg1b表达量在4-6d时显著降低(p<0.05)。说明转基因菌株在侵染过程中有sirna的释放。用转基因菌株孢子悬浮液处理柑橘木虱成虫10d后,通过计算lc50值和lt50值评估菌株对柑橘木虱成虫的毒力。一、具体方法步骤为:
1.转基因菌株对柑橘木虱的毒力
用吸虫器在田间吸取柑橘木虱成虫后,在室内九里香苗子上饲养1d,以便柑橘木虱成虫适应新的寄主。对柑橘木虱成虫侵染的处理为用转基因渐狭蜡蚧菌孢子悬浮液浸没处理柑橘木虱雌成虫15s,每处理选择30头发育一致,活泼健康的柑橘木虱成虫于装有新鲜九里香枝条的玻璃瓶中,用保鲜膜封口,在保鲜膜上扎上均匀细密的小孔。放入温度27±0.5℃、湿度80%±5%、14l:10d的恒温光照培养箱中。自3d后每24h调查死亡率至10d。将每天调查的尸体从玻璃瓶中挑出,放置在铺有湿润的洁净滤纸的一次性培养皿中,将培养皿放入同一个光照培养箱培养。待虫体上长出白色菌丝后,菌株重新分离验证,如验证成功即表示侵染成功。统计侵染率。每处理设置3个重复,以0.05%吐温-80溶液处理作对照。
2.数据处理
转基因菌株对柑橘木虱雌成虫的毒力测定结果用spss24.0分别对剂量和时间作线性回归分析,计算出剂量效应lc50和时间效应lt50,并且用duncan多重比较法进行方差分析。所得结果均用excel2016作图。
二、结果
结果如表3、4所示,发现la::dcvg1a和la::dcvg1b菌株的lc50分别为1.10×104和5.60×104孢子/ml,均显著低于la::gfp菌株lc50(1.60×106);la::dcvg1a、la::dcvg1b菌株的lt50分别为4.67d、4.33d,均显著短于野生型菌株(wt)5.22d(p<0.05)。结果表明,转基因渐狭蜡蚧菌la::dcvg1a和la::dcvg1b可有效提高其对柑橘木虱毒力的毒力,缩短对木虱的浸染时间。
表34株渐狭蜡蚧菌(10d)对柑橘木虱成虫的致死中浓度
注:lc50值为平均值和置信区间,小写字母表示单因素方差分析(p<0.05)结果。
表44株渐狭蜡蚧菌(1.00×108孢子/ml)对柑橘木虱成虫的致死中时
序列表
<110>西南大学
<120>干扰柑橘木虱卵黄蛋白原基因表达的转基因生防真菌及其制备方法和应用
<160>20
<170>patentinversion3.5
<210>1
<211>999
<212>dna
<213>人工序列
<400>1
gcatggaaacaaggacaagtctatgaataccaaatccaaggccgcaccctagctgctctt60
catgatgtagctgaccaatacaccggaaccatcatcaaagccaccctcaaggttcaaccc120
agaaatcaagactcagtcttggcttgggtcaccaacgctagacactctgatgttcacgcc180
aacctaaccaatggctggaaccaagagatcccagacaaatacctcaactaccaaaactgg240
caactcagtgacaaaccattcgccatccaattcaagaatggagttgttaagaatctgcaa300
gtcagccaaaacctgccaacctgggaactgaacatcatcaagagtattgtcagtcaactc360
caagttgacaccagagccgaaaatgaagtcagctcgcgtctcaaccagaaacccaagaac420
ggcaaacctttcggaaccttcaagaccatggaagacaccgtcactggagaatgtgaaacc480
ttgtacgacatcaaacctctgcaacaagttgaacaacaaaacaaacctcaactcgccccc540
atgccaaacctcaagggatcaaacggagaccttattgacatcatcaagaccaagaacttc600
agtcgttgtgactccagaatcgaataccacttcggacttcccggaagcaacgatgttgaa660
ccatcaagcaaccaagttcttaacttcttgtccagatcttcaaccagcagagtcatcatt720
gctggagacctgagccactacaccatccaatcctcagtcaccaccgacaaaattgtcatc780
agtcccgaactctacaacaaacaaaagggaatggttgtcagtcgcctgaacgttactctg840
tccaacgtccactctgctgctcaaaacaatgccccagctctcccatccaacgtcaacaaa900
gttgatgatttgatctacgaatacaacccagcttcccctgacaacgaatctgcccaaaac960
aacaacggcaagaacaaccaccacaacgacgatgactca999
<210>2
<211>969
<212>dna
<213>人工序列
<400>2
gctgtcaccaaatctttggcccaacaaattggtcaagaaatccaagaacccaacaccttg60
gctgaacacaaaaccctttccaaattcaccatcctcgccggagttatccgcaccatgaac120
gccaaacaactggaagccgccactcacgaactctactaccaacaaaacaaggcctcttca180
tccagccaatccgatgccaccaaattaaccgcatggaaagcttaccgtgacgccgttgct240
caagcaggaactggtcccgctttgatggccatgaaacaatggattgaatccggcaaagtc300
gaaggagaagaagctgctgaattattggctgttctccccaacaccgccagatacccaacc360
cgtgaatacatcaaggaattcttcaaccttgccaccagctctcaagtcaccaaacaagct420
cacctcaacacctctgctatcctctccgtcgccagtttggcaagaaaagcccaagttgac480
tctgacaactcacacaaccaatacccagttcatgctttcggacccctgtcatccaaaaac540
agcaaagacatcactgaaagatacatcccatacttggccaacaaactcaaggaagctacc600
agaaaccaagacagtctgaaagctcaagtctacatcaaagctctcggaaacttaggacat660
accgccgtcctcgctgtcttcaaaccataccttgaaggaaaagccccagccaccaacttc720
caacgtctttcaatggttgccgccatggaccaagttgccagattatcaccaaaatctgtc780
caacctgctctcttcaacatctacctcaacaccggagaaagccacgaattacgttgtgct840
gctgtattccaactcatgaagacctacccatccgctcaattgctccaacgtatggctgca900
ttcactgaacaagacatgagcaaacaagttaactctgctgtcaaatccgccatcgaaagc960
gctgctgaa969
<210>3
<211>48
<212>dna
<213>人工序列
<400>3
tactgaattaacgccgaattaattcggatgcttgggtagaataggtaa48
<210>4
<211>40
<212>dna
<213>人工序列
<400>4
atagagtagatgccgctgcagatggtgagcaagggcgagg40
<210>5
<211>46
<212>dna
<213>人工序列
<400>5
tctacccaagcatccctcgaggcatggaaacaaggacaagtctatg46
<210>6
<211>45
<212>dna
<213>人工序列
<400>6
aagaaattcttacacatttaaattgagtcatcgtcgttgtggtgg45
<210>7
<211>46
<212>dna
<213>人工序列
<400>7
ggtcttaattaactctctagagcatggaaacaaggacaagtctatg46
<210>8
<211>43
<212>dna
<213>人工序列
<400>8
aatttgcaggtatttggatcctgagtcatcgtcgttgtggtgg43
<210>9
<211>42
<212>dna
<213>人工序列
<400>9
tctacccaagcatccctcgaggctgtcaccaaatctttggcc42
<210>10
<211>42
<212>dna
<213>人工序列
<400>10
aagaaattcttacacatttaaatttcagcagcgctttcgatg42
<210>11
<211>42
<212>dna
<213>人工序列
<400>11
ggtcttaattaactctctagagctgtcaccaaatctttggcc42
<210>12
<211>40
<212>dna
<213>人工序列
<400>12
aatttgcaggtatttggatccttcagcagcgctttcgatg40
<210>13
<211>22
<212>dna
<213>人工序列
<400>13
ctggatgcttgggtagaatagg22
<210>14
<211>23
<212>dna
<213>人工序列
<400>14
caaaggaatagagtagatgccgc23
<210>15
<211>21
<212>dna
<213>人工序列
<400>15
agcagacaggaacgaggacat21
<210>16
<211>21
<212>dna
<213>人工序列
<400>16
accccaacccaaaaagagtgt21
<210>17
<211>20
<212>dna
<213>人工序列
<400>17
cttgccttggagtttatgtt20
<210>18
<211>21
<212>dna
<213>人工序列
<400>18
ttattcttgttgacatggagc21
<210>19
<211>20
<212>dna
<213>人工序列
<400>19
ctacacagccatcggtccag20
<210>20
<211>20
<212>dna
<213>人工序列
<400>20
gttcgcccttcctcccttta20
- 还没有人留言评论。精彩留言会获得点赞!