一种诱导型碱基编辑系统及其应用
本发明属于基因工程技术领域,具体涉及一种诱导型碱基编辑系统及其应用。
背景技术:
碱基编辑是一种基因组编辑策略,可直接在基因组dna中转换特定的单个核苷酸(reesandliu2018)。目前,碱基编辑器有两种类型,胞嘧啶碱基编辑器(cbe)和腺嘌呤碱基编辑器(abe),两者都由脱氨酶与催化缺陷的cas9蛋白组成,该酶可催化胞嘧啶或腺嘌呤的脱氨反应。在sgrna的指导下,碱基编辑器可以在靶点内特定窗口的几个碱基对中将c-g碱基对转换为t-a碱基对,或将a-t碱基对转换为g-c碱基对。因为它高效且精确,并且不会产生由dna双链断裂(dsb)引入的插入或缺失等副产物,所以该技术很快被广泛用于各种模式生物中,例如小鼠,斑马鱼,拟南芥和酿酒酵母。
尽管碱基编辑技术在广泛的基础研究中取得了许多令人振奋的成果,但是碱基编辑在临床应用中仍然存在一个主要阻碍,即在dna或rna水平上的脱靶效应。当前的cbe和abe通常会引起rna中全转录组范围内的脱靶,引起了对安全性问题的关注。更严重的是,由于cbe还会造成dna水平的脱靶,可能进一步导致包括癌症在内的病理状况。实际上,早在三十多年前,研究人员发现过表达apobec1(cbe中使用的最流行的胞嘧啶脱氨酶之一)可导致转基因小鼠患上肝癌。随后,aid也被发现在小鼠中过表达会诱导肿瘤的发生。基于这些发现,越来越多的研究证实了胞嘧啶脱氨酶与多种组织中的肿瘤发生之间的联系。已发现的胞嘧啶脱氨酶的成员中,如apobec3b,在多种实体瘤。
已发现胞嘧啶脱氨酶的过表达能够诱导基因组dna中的碱基替换,从而损害基因组稳定性。更重要的是,已发现许多肿瘤细胞带有以胞嘧啶标记为特征的snv。因此,不受控制的碱基编辑器(尤其是cbe)可能会产生肿瘤。考虑到aav是目前最有效的体内传递载体之一,aav注射后会使转基因在体内长期表达,使用这种载体传递碱基编辑器无疑会放大脱靶后果。因此,我们迫切需要探索活性可控的新型碱基编辑工具。
技术实现要素:
针对现有技术中的上述不足,本发明提供一种诱导型碱基编辑系统及其应用,通过构建拆分式碱基编辑系统,并能通过雷帕霉素的诱导来控制其碱基编辑活性,通过该方案能够有望缩短编辑时间。同时,本申请中的拆分设计不会减少目标编辑,因此,可以作为蛋白质突变的补偿策略,并且可以与这些突变的脱氨酶结合使用,以进一步减少脱靶编辑。
近年来,许多研究表明碱基编辑器存在的dna和rna的脱靶效应。本发明通过控制脱氨酶活性进而控制碱基编辑器的编辑编辑活动,从而降低脱靶效应。本发明的碱基编辑系统包括可诱导的人源胞嘧啶脱氨酶(a3a)与单切口活性的spcas9。具体方案为:将a3a分裂为失活的n端和c端后,分别与frb和fkbp蛋白融合,frb和fkbp在雷帕霉素诱导下形成稳定的三元复合物,从而使脱氨酶重新组装成有功能性的a3a。由于脱氨酶的活性可受诱导剂调控,进而该碱基编辑器的活性是可控的。
为实现上述目的,本发明解决其技术问题所采用的技术方案是:
一种诱导型碱基编辑系统,碱基编辑系统包括碱基编辑器,以及与其结合的雷帕霉素;碱基编辑器包括脱氨酶;
脱氨酶能基于任一氨基酸位点进行拆分;雷帕霉素通过拆分后的氨基酸位点与脱氨酶结合,并诱导完成碱基编辑。
进一步地,碱基编辑器为由脱氨酶和be3/spcas9组成的碱基编辑器。
进一步地,雷帕霉素的frb和fkbp分别与脱氨酶氨基酸拆分位点的非结构性环的分裂处偶联。
进一步地,frb和fkbp分别与脱氨酶氨基酸拆分位点的n端和c端融合,并二聚化形成异二聚体,完成碱基编辑器的组装。
进一步地,脱氨酶的拆分位点为第44位、85位、118位或147位氨基酸。
进一步地,脱氨酶的拆分位点为第85位氨基酸。
进一步地,脱氨酶为胞嘧啶脱氨酶。
进一步地,胞嘧啶脱氨酶为apobec3a胞嘧啶脱氨酶或apobec1胞嘧啶脱氨酶。
进一步地,雷帕霉素的浓度为0.01~200nm。
进一步地,雷帕霉素的浓度为200nm。
上述诱导型碱基编辑系统在基因编辑中的应用。
一种用于进行基因编辑的试剂盒,包括上述诱导型碱基编辑系统。
本申请构建得到的碱基编辑系统能够使得cas9保持完整,因此它能与其他非传统的碱基编辑器兼容,例如cp-cas9衍生的碱基编辑器(huang等,2019),内部镶嵌的be-pigs碱基编辑(wang等人,2019)和sgrna骨架修饰的sgbes(wang等人,2020)。
本发明的有益效果为:
本申请通过构建拆分式碱基编辑系统,并能通过雷帕霉素的诱导来控制其碱基编辑活性,通过该方案能够有望缩短编辑时间。同时,本申请中的拆分设计不会减少目标编辑,因此,可以作为蛋白质突变的补偿策略,并且可以与这些突变的脱氨酶结合使用,以进一步减少脱靶编辑。
附图说明
图1为诱导型split-a3a-be3的设计流程图;
图2为4个split-a3a-be3的可诱导编辑活性检测结果;
图3为雷帕霉素的作用浓度和持续时间对sa3a-be3-85的碱基编辑的影响检测结果;
图4为sa3a-be3-85的编辑模式的特征检测;
图5为sa3a-be3-85的dna脱靶编辑检测结果;
图6为通过ert2系统控制sa3a-be3-85的亚细胞定位减少背景编辑的检测结果;
图7为split-apobec1-be3的构建过程和特性检测。
具体实施方式
下面对本发明的具体实施方式进行描述,以便于本技术领域的技术人员理解本发明,但应该清楚,本发明不限于具体实施方式的范围,对本技术领域的普通技术人员来讲,只要各种变化在所附的权利要求限定和确定的本发明的精神和范围内,这些变化是显而易见的,一切利用本发明构思的发明创造均在保护之列。
实施例1构建split-a3a质粒
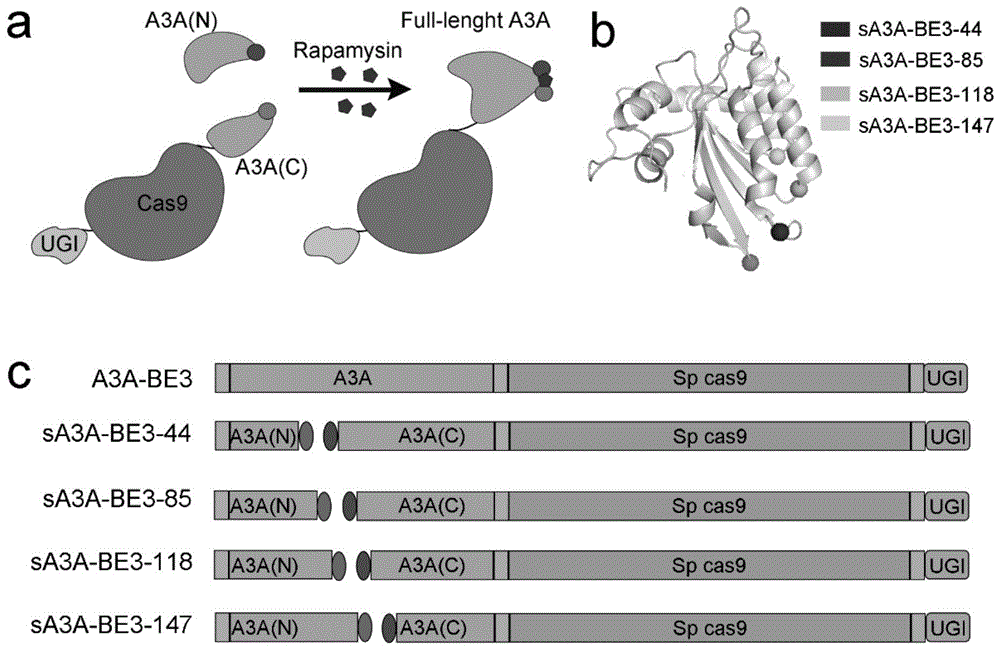
在a3a-be3的基础上中构建了split-a3a质粒,以形成一系列的n端split-a3a及其对应的c端split-be3-a3as(图1b)。如图1c所示,构建了4组split-a3a-be3(sa3a-be3),并以其氨基酸拆分位点命名,即sa3a-be3-44,sa3a-be3-85,sa3a-be3-118和sa3a-be3分别为-147。
图1为诱导型split-a3a-be3的设计;其中,图a显示了诱导型split-a3a-be3重组装示意图,雷帕霉素诱导分别与a3a的n端和c端融合的frb和fkbp的二聚化从而形成异二聚体,从而完成功能性a3a-be3碱基编辑器的组装。图b为a3a结构的示意图(pdb:5keg)。与ssdna-a3a结合界面相对的四个非结构性环是候选的分裂位点,在图中以球状表示。图c为split-a3a-be3胞嘧啶碱基编辑器的构建示意图。
实施例2split-a3a碱基编辑
1、sa3a-be3可诱导的碱基编辑活性检测
在3个内源性靶点中,将每对split-a3a-be3编辑器分别与sgrna共转染到hek293t细胞中。如图2a所示,所有sa3a-be3都表现出可诱导的编辑活性,尽管具有不同的潜力。在这些sa3a-be3中,当用200nm雷帕霉素诱导时,sa3a-be3-44在所有3个靶位点上的编辑效率最高,该浓度在frb/frbp的split系统中经常使用。然而,其活性在未诱导条件下基本保持不变。平均而言,未诱导的sa3a-be3-44表现出与全长a3a-be3相当的活性(图2b)。sa3a-be3-44的c末端部分的转染未产生任何可检测的编辑,这表明sa3a-be3-44的背景编辑活性不是由于c末端a3a的残留脱氨活性,可能是由于脱氨酶的自组装造成的。相反,sa3a-be3-147在未诱导和诱导条件下均表现出最小的编辑活性,分别为全长a3a-be3活性的约7%~19%(图2b)。sa3a-be3-85和sa3a-be3-118比上述两者更有效地被诱导。平均而言,在诱导条件下,sa3a-be3-85的活性与全长a3a-be3相似,而sa3a-be3-118的活性则比全长低约1.7倍。在所测试的四个split-a3a-be3碱基编辑器中,sa3a-be3-85表现出对雷帕霉素的最佳响应效果(图2b)。
图2为4个split-a3a-be3的可诱导编辑活性;其中,图a为诱导型碱基编辑器的编辑效率测定,在3个内源性靶点中,全长的a3a-be3或诱导型的sa3a-be3s的质粒分别与sgrna共转染至hek293t细胞。通过sanger测序和editr(kluesneretal.2018)分析碱基编辑效率。每个实验至少重复三遍。数据表示为平均值±sem。图b为图a中所有编辑器在每个靶点的平均编辑效率。以全长a3a-be3的编辑效率为1做归一化处理。
2、sa3a-be3-85表征
检测雷帕霉素的诱导时间对编辑效率的影响。将上述用sa3a-be3-85编辑器转染的hek293t细胞分别用200nm雷帕霉素处理3、6、12、24和48小时。如图3a,c所示,我们观察到具有时间依赖性的的编辑。有趣的是,在测试的三个目标位点中,24小时诱导的编辑效率与两个位置的48小时诱导相似,而对于其余一个位点,24小时诱导的编辑效率比48小时诱导的编辑效率低40.36%,这表明诱导持续时间的影响是靶标特异性的。接下来,我们测试了雷帕霉素的浓度对编辑效率的影响,用浓度范围从0.01nm至200nm的雷帕霉素处理转染的细胞(图3b)。如预期的那样,观察到剂量依赖性效应,并且剂量效应曲线显示在50nm左右开始达到平台期。(图3d)。
图3为雷帕霉素的作用浓度和持续时间对sa3a-be3-85的碱基编辑的影响;其中,图a为sa3a-be3-85在200nm雷帕霉素处理下不同诱导时间的编辑效率。用200nm雷帕霉素处理转染的hek293t细胞,并在相应的时间点收细胞。图b为不同浓度雷帕霉素诱导48小时后sa3a-be3-85的编辑效率。用各种浓度的雷帕霉素处理转染的hek293t细胞48小时,然后进行序列分析。通过平均分析每个位点所有可靶向胞嘧啶的编辑效率,总结了诱导持续时间(图c)和强度(图d)对sa3a-be3-85碱基编辑的影响。
实施例3split-a3a的进一步表征
检测第85氨基酸处a3a的分裂是否改变了a3a-be3的碱基编辑模式,包括编辑窗口,序列偏好性和c→t的编辑纯度。
为了确定编辑窗口,选择了3个在编辑窗口内具有多个胞嘧啶的靶点(图4a)。如图4a所示,与全长a3a-be3相比,sa3a-be3-85不会明显移动编辑窗口的位置或影响编辑窗口的宽度。然后,我们测试了sa3a-be3-85是否具有与全长a3a-be3不同的序列偏好性,因为先前的研究表明脱氨酶螺旋之间的非结构化环在底物识别中起着重要的作用(salteretal.2016)。根据ncmotif(gc,cc,ac和tc)将9个靶点分为四个组,并比较了sa3a-be3-85和全长a3a-be3的序列偏好性(图4b)。平均而言,sa3a-be3-85在所有四种不同motif中的编辑效率与全长a3a-be3相似。sa3a-be3-85占全长a3a-be3的编辑效率为相对编辑效率,对于gc为84.5%,对于cc为99.7%,对于tc为93.5%,对于ac为95.0%,这表明sa3a-be3-85并没有明显改变的基序偏好。为了测试c到t的编辑纯度,我们选择了2个先前表征的位点,除了c到t之外,这些位点还倾向于从c转换为g(hek2中的c6和rnf2中的c6)。我们发现sa3a-be3-85的编辑纯度比原始a3a-be3高53.05%,在rnf2站点高9.18%,这表明sa3a-be3-85稍微提高了c→t产品的纯度,具有更高的c→t效率(图4c)。
图4为sa3a-be3-85的编辑模式的特征;其中,图a为sa3a-be3-85和全长a3a-be3的编辑窗口比较;图b为sa3a-be3-85和全长a3a-be3的序列偏好性比较,计算每个靶点中各种ncmotif的碱基编辑效率;图c为全长a3a-be3和sa3a-be3-85的编辑纯度比较。
实施例4split-a3a-bes的dna脱靶编辑
cbe诱导序列依赖性和非序列依赖性脱靶编辑的事实大大限制了其应用。为了确定sa3a-be3-85的序列依赖性脱靶编辑,选择了两个特征明确的靶标(heksite4和exmi)。如图5a所示,sa3a-be3-85通常在所有三个脱靶位点上均显示出较低的序列依赖性脱靶编辑。随着诱导时间的延长,sa3a-be3-85在脱靶位点的编辑效率逐渐提高(图5a)。计算了编辑效率比脱靶效率的比值,发现sa3abe3-85(4:1)高于a3a-be3(2.4:1)(图5b)。
然后通过正交r环测定法测试了sa3a-be3-85的非序列依赖性的脱靶编辑,人工r环通过在特定基因组位点上转染单切口酶sacas9和sgrna诱导形成。碱基编辑器与r环构建体的共转染显示,与全长a3a-be3相比,sa3a-be3-85显示出脱靶编辑显着减少(图5c)。以上结果表明,与全长a3a-be3相比,sa3a-be3-85在序列依赖性和非序列依赖性的dna脱靶编辑中都显示出较低的倾向。
图5为sa3a-be3-85的dna脱靶编辑,其中,图a为sa3a-be3-85的序列依赖性脱靶编辑效率,分别靶向hek4和emx1位点的两个grna用于分析依赖序列的脱靶编辑,脱靶位点通过序列相似性进行预测,并已在(komoretal。2016)中进行了描述。在每个位点分析所有可靶向胞嘧啶的平均编辑效率;图b为目标编辑与脱靶编辑比值计算的相对编辑效率;图c显示了通过正交r环检测的原理及sa3a-be3-85的非序列依赖性的脱靶编辑检测。通过转染nsacas9和相应的sgrna产生了两个人工r环,同时测定了其目标编辑效率(exm1)
实施例5通过控制亚细胞定位减少split-a3a的背景编辑
由于split-a3a-85在不受雷帕霉素诱导时显示出背景编辑,因此我们试图测试是否可以通过调节split-a3a-85组分的亚细胞定位来进一步减少背景编辑,从而使它们在无需诱导时在空间上分离。
使用核转运系统ert2来控制split-a3a-85的n端部分的核酸转运,以便n端split-a3a-85位于胞浆中,并在空间上与其c端隔开,从而阻断自动组装(图6a)。如图6b所示,ert2结构域与n-分裂-a3a-85的n-末端而非c-末端的融合在诱导下实现了诱导编辑,并且显着减少了无诱导时的背景编辑。
图6用ert2系统控制sa3a-be3-85的亚细胞定位可减少背景编辑;其中,图a显示了用ert2系统控制sa3a-be3-85的核转运的机制,sa3a-be3-85的n端部分与ert2融合,并被4-oht诱导转移到核中,在此与sa3a-be3-85的c端部分靠近,并在存在雷帕霉素的情况下组装功能性碱基编辑器;图b为4-oht和雷帕霉素双重控制系统诱导的sa3a-be3-85构建体的构建;图c为融合ert2的split-a3a-be3-85的背景编辑测定。
实施例6大鼠apobec1衍生的碱基编辑器的设计和表征
建立split-a3a-be3系统后,我们接下来试图将这一发现扩展到另一个广泛使用的胞嘧啶脱氨酶rapobec1,该酶也经常在碱基编辑中使用。通过序列比对,rapobec1与a3a有48.1%的相似性,并且负责催化脱氨的关键氨基酸在这两个胞嘧啶脱氨酶之间基本保持保守。由于rapobec1的晶体结构尚未被解析,因此我们使用在线程序(mpibioinformaticstoolkit:https://toolkit.tuebingen.mpg.de。)根据蛋白质序列预测其结构。rapobec1的预测结构与a3a相似,特别是a3a中的环4和附近结构与rapobec1中的相应区域高度同源。如图7a,b所示,我们通过结构比较和序列比对将a3a的第85个氨基酸映射到rapobec1的第77个氨基酸。所得的分裂-rapobec1-be3在诱导条件下达到了全长be3的85.83%的活性,而其活性却显着降低而没有受到干扰,这与sa3a-be3-85的先前结果一致(图7c)。因此,该结果表明我们的拆分策略也可以应用于其他脱氨酶。
图7为split-apobec1-be3的设计和特性;其中,图a为apobec3a和apobec1的氨基酸序列比对;图b为a3a和apobec1的结构比较,mpi生物信息学工具包预测了apobec1的结构,红色区域显示了分裂位点;图c为split-apobec1-be3的诱导活性检测。
本申请通过构建拆分式碱基编辑系统,并能通过雷帕霉素的诱导来控制其碱基编辑活性,通过该方案能够有望缩短编辑时间。同时,本申请中的拆分设计不会减少目标编辑,因此,可以作为蛋白质突变的补偿策略,并且可以与这些突变的脱氨酶结合使用,以进一步减少脱靶编辑。
除此之外,本申请构建得到的碱基编辑系统能够使得cas9保持完整,因此它能与其他非传统的碱基编辑器兼容,例如cp-cas9衍生的碱基编辑器(huang等,2019),内部镶嵌的be-pigs碱基编辑(wang等人,2019)和sgrna骨架修饰的sgbes(wang等人,2020)。
- 还没有人留言评论。精彩留言会获得点赞!