聚酯酰胺、聚酯酰胺纤维及制备方法
1.本发明涉及一种聚酯酰胺、聚酯酰胺纤维及制备方法。
背景技术:
2.聚对苯二甲酸乙二醇酯(pet)具有苯环,分子模量高,整个pet分子链表现为刚性,具有良好的力学性能和尺寸稳定性。然而,由于pet大分子的结晶度高,缺少亲水性基团,因此采用pet制成的纤维吸湿性、染色性较差,纤维回潮率仅为0.4%左右,织物手感较差。
3.国内外现有围绕聚酯纤维吸湿改性的方法主要有共混改性、制成纤维截面异性结构、表面涂覆/整理、共聚改性等,前面三种改性方法分别存在相容性差、吸湿性提高不大、改性效果不持久等问题,纺丝效果不理想。此外,由于pet纤维的染色性能差,为了实现较高的上染率,常常需要在较高的温度(例如130℃左右)条件下进行着色,而高温操作一般比较危险,对染色设备要求较高,且浪费能源;并且即使在高温条件下染色,也有待进一步提高。
4.因此,亟需提高pet亲水性和柔顺性,同时能够改善pet纤维的染色性能、尤其低温条件的染色性能。
技术实现要素:
5.本发明要解决的技术问题是克服现有技术中pet亲水性和柔顺性较差,以及pet纤维的上染率、尤其是较低温条件的上染率不高的缺陷,而提供一种聚酯酰胺、聚酯酰胺纤维及制备方法。本发明的聚酯酰胺亲水性和柔顺性较高,对纤维纺制设备较为友好;所制得的聚酯酰胺纤维染色性能好,并且能够在较低温度下实现较好的染色效果,同时条干不均率低。
6.本发明是通过下述方案来解决上述技术问题的:
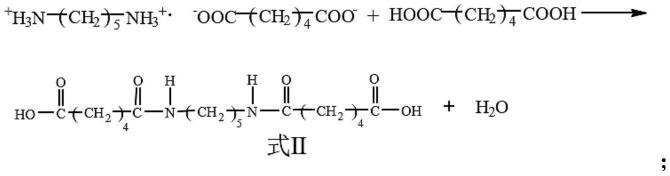
7.本发明提供一种聚酯酰胺的制备方法,其包括如下步骤,将如式ⅱ所示的pa56盐衍生物与对苯二甲酸乙二醇酯(bhet)进行缩聚反应,即得;其中,所述缩聚反应的温度为235~280℃。
[0008][0009]
本发明中,所述pa56盐衍生物分子式为c
17h30
o6n2,相对分子质量为358。
[0010]
本发明中,所述缩聚反应的温度较佳地为250~270℃,进一步较佳地为250~260℃,更佳地为250~255℃。所述缩聚反应的其他条件可为本领域常规。所述缩聚反应的压力一般为真空度50pa以下。所述缩聚反应的时间可为2.5~3.5h,较佳地为3~3.5h。
[0011]
本发明中,所述pa56盐衍生物与所述对苯二甲酸乙二醇酯的摩尔比可为本领域常规,例如可为(0.04~0.30):1,再例如0.05:1.1、0.1:1.4、0.15:1.3或者0.2:1.2,较佳地为(0.07~0.15):1,进一步较佳地为(0.07~0.12):1,更佳地为(0.10~0.12):1。
[0012]
本发明中,所述pa56盐衍生物可市售可得或者按照本领域常规方式制得。较佳地,
所述pa56盐衍生物按照如下步骤制得:将pa56盐与己二酸进行反应即可;
[0013][0014]
其中,所述pa56盐与所述己二酸的摩尔比可按照本领域反应的常规摩尔比,较佳地为1:(1~1.6),例如1:1.4。所述pa56盐与所述己二酸反应的温度较佳地为145~165℃,例如150℃或者160℃。所述pa56盐与所述己二酸反应的时间可为10~20h,较佳地为10~18h,例如14h。
[0015]
其中,所述聚酯酰胺的制备方法一般还包括步骤(a)分离的步骤与步骤(b)提纯的步骤。
[0016]
步骤(a)的分离与步骤(b)的提纯的步骤和条件可按照本领域常规。所述分离的步骤可包括共沸精馏分离得到含pa56盐衍生物的混合物。所述共沸精馏采用的共沸剂可包括二甲苯和/或三甲苯。所述共沸精馏的温度为148℃~160℃例如150℃或者160℃。所述共沸精馏的时间为12~20h,例如15h或者19h。所述共沸精馏的装置可采用油水分离装置。较佳地,所述pa56盐和所述己二酸边的反应与所述共沸精馏同步进行。所述共沸剂与所述pa56盐的体积质量比可为(6~8):1(ml/g),例如7:1(ml/g)。
[0017]
所述提纯的步骤可包括:将分离所得含pa56盐衍生物的混合物与醇类溶剂混合、经二次沉淀,即得含pa56盐衍生物的滤液。所述含pa56盐衍生物的混合物与所述醇类溶剂的混合温度可为62~68℃,例如65℃。所述二次沉淀中的第一次沉淀的温度可为常温。所述二次沉淀中的第二次沉淀的温度可为0~10℃。所述醇类溶剂可为乙醇。所述提纯的次数较佳地为1~5次,例如3次。一般地,所述提纯之后还包括将所述含pa56盐衍生物的滤液进行干燥。所述干燥的方式可为旋转蒸发。
[0018]
本发明中,所述对苯二甲酸乙二醇酯可市售可得或者按照本领域常规方式制得。较佳地,所述对苯二甲酸乙二醇酯按照如下步骤制得:将对苯二甲酸与乙二醇进行酯化反应即可;
[0019][0020]
其中,较佳地,所述对苯二甲酸(pta)与所述乙二醇(eg)的摩尔比为1:(1.2~1.6),例如1:1.4。
[0021]
其中,所述酯化反应的温度可为225~248℃,例如240℃,较佳地为230~237℃。所述酯化反应的时间较佳地为2~3h。
[0022]
其中,所述酯化反应的催化剂可为三醋酸锑、三氧化二锑、乙二醇锑中的一种或多种。
[0023]
本发明中,所述缩聚反应体系还可包括如下添加剂,例如抗氧剂和/或光稳定剂。
所述抗氧剂可包括本领域常规的抗氧剂,例如所述抗氧剂包括抗氧剂1098、抗氧剂168、二异丙基苯基二苯胺、抗氧剂n,n'-双(2,2,6,6-四甲基-4-哌啶基)-1,3-苯二甲酰胺、次磷酸钠、苯基次磷酸钠和磷酸三甲酯中的一种或者多种。所述光稳定剂可包括本领域常规的光稳定剂,例如所述光稳定剂包括氧化锌、钛白粉r103、光稳定剂bw-10ld和群青5008中的一种或者多种。所述抗氧剂的加入量为所述聚酯酰胺的0.001wt%~0.1wt%。所述光稳定剂的加入量为所述聚酯酰胺的0.001wt%~5wt%。
[0024]
在本发明一较佳实施例中,所述pa56盐衍生物与所述对苯二甲酸乙二醇酯的摩尔比为(0.10~0.12):1,所述缩聚反应的温度为250~255℃,所述缩聚反应的时间为3~3.5h,所述缩聚反应的压力为真空度控制在50pa以下。
[0025]
在本发明一更佳实施例中,所述pa56盐衍生物与所述对苯二甲酸乙二醇酯的摩尔比为0.15:1.3,所述缩聚反应的温度为255℃,所述缩聚反应的时间为3.5h,所述缩聚反应的压力为真空度控制在50pa以下。
[0026]
本发明中,将所得到的聚酯酰胺按照本领域常规的方式进行预结晶,例如将聚酯酰胺切粒在132~137℃预结晶1~3h后,利用真空转鼓在140~147℃下干燥10~30小时,得干燥切片,铝塑袋密封保存。
[0027]
本发明还提供一种聚酯酰胺,其由上述制备方法制得。
[0028]
本发明中,所述聚酯酰胺的粘均分子量较佳地为18000~22600,更佳地为18070~22570,例如18750、20120、22570或者20470。
[0029]
本发明中,所述聚酯酰胺的结晶度较佳地为24~34%,例如33.97%、32.73%、28.97%或者26.88%,更佳地为24~29%。所述结晶度的测试条件可为辐射靶源为cu靶,波长为15.4nm,电压为40kv,扫描范围为5
°
~60
°
。
[0030]
本发明中,所述聚酯酰胺的玻璃化转变温度(tg)较佳地为45~65℃,例如64.5℃、59.1℃、48.4℃或者46.9℃,更佳地为45~49℃。
[0031]
本发明中,所述聚酯酰胺的特性粘度较佳地为0.65~0.78dl/g,例如0.67dl/g、0.71dl/g、0.78dl/g或者0.72dl/g。
[0032]
本发明中,所述聚酯酰胺的静态接触角较佳地为65~85
°
,例如82.3℃、77.4℃、73.6℃或者68.1℃,更佳地为65~74
°
。
[0033]
本发明还提供一种聚酯酰胺纤维的制备方法,其包括如下步骤:将上述聚酯酰胺制成聚酯酰胺纤维。
[0034]
本发明中,所述聚酯酰胺制成聚酯酰胺纤维的方式可按照本领域常规的制备纤维的方法,例如将所述聚酯酰胺进行熔融、纺丝即可;再例如还包括牵伸热定型的步骤。较佳地,将所述聚酯酰胺在螺杆挤压机中进行熔融、纺丝即得预取向丝,再将所述预取向丝进行牵伸热定型,得到聚酯酰胺纤维。
[0035]
其中,所述熔融的加热方式可为本领域常规,例如采用纺丝螺杆挤压机进行五区加热方式。所述五区加热方式中,一区温度可为248~255℃,例如250℃。二区温度可为270~275℃,例如272℃。三区温度可为278~283℃,例如280℃。四区温度可为280~285℃,例如282℃。五区温度可为275~280℃,例如278℃。
[0036]
其中,所述纺丝的条件可为本领域常规。所述纺丝的温度可为270~285℃,例如278℃。所述纺丝过程的压力可为10~15mpa,例如12mpa。所述纺丝速度可为2500~4000m/
min,例如3500m/min。
[0037]
其中,所述牵伸热定型的步骤和条件可为本领域常规。所述牵伸热定型的温度可为125~135℃,例如130℃。所述牵伸热定型的牵伸倍数可为1.2~1.7倍,例如1.5倍。
[0038]
本发明还提供一种聚酯酰胺纤维,其由上述制备方法制得。
[0039]
本发明还提供一种聚酯酰胺纤维,所述聚酯酰胺纤维在100~130℃的上染率为80%以上。
[0040]
本发明中,所述聚酯酰胺纤维在100~130℃温度下的上染率更佳地为83~99.5%,例如83.2%、85.6%、98.7%、99.2%、98.8%、99.1%、99.2%或者99.4%。进一步更佳地,在100℃时的上染率为83~99.5%。进一步更佳地,在130℃时的上染率为98.5~99.5%。
[0041]
本发明中,所述聚酯酰胺纤维的染色m率较佳地为99%以上,例如99.2%、99.6%或者99.7%。
[0042]
本发明中,所述聚酯酰胺纤维的条干不均率较佳地为1~2%,例如1.28%或者1.92%,更佳地为1.28%。
[0043]
本发明中,较佳地,所述聚酯酰胺纤维包括poy纤维、fdy纤维或者dty纤维。
[0044]
本发明中,较佳地,所述聚酯酰胺纤维由上述聚酯酰胺纤维的制备方法制得。
[0045]
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0046]
本发明所用试剂和原料均市售可得。
[0047]
本发明的积极进步效果在于:
[0048]
1)本发明聚酯酰胺的tg较低,均在65℃以下,易于上染,且分子链的柔顺性较好。
[0049]
2)本发明聚酯酰胺的接触角均低于90℃,亲水性较好。
[0050]
3)本发明的聚酯酰胺纤维的染色性能较好、尤其是在较低温度下还能实现较好的染色效果;染色m率较高。
[0051]
4)本发明的聚酯酰胺纤维的条干不均率较低。
附图说明
[0052]
图1为对比例1和实施例3~6聚酯酰胺的1h-nmr图谱。
[0053]
图2为图1中的区域a的放大图谱
具体实施方式
[0054]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0055]
实施例1pa56盐衍生物的制备
[0056]
采用己二酸对pa56盐进行封端处理,反应式如下,pa56盐衍生物的分子式为c
17h30
o6n2,相对分子质量为358。
[0057][0058]
a、将pa56盐、己二酸与二甲苯混合后进行反应,边反应边进行共沸蒸馏除去生成的水,制得含有pa56盐的衍生物的混合物;其中,pa56盐与二元酸的摩尔比为1:1.4,二甲苯的体积与pa56盐的质量之比为7:1(ml/g),反应温度为160℃,反应时间为20h,共沸蒸馏采用油水分离装置,共沸蒸馏的温度为160℃,共沸蒸馏的时间为19h;
[0059]
b、反应结束后使用醇类有机溶剂(乙醇)进行提纯,具体过程为:首先将反应后的混合物在65℃的温度条件下与醇类有机溶剂混合,冷却至常温、进行第一次沉淀,沉淀出不溶的反应物,过滤后得到第一过滤液,然后将过滤液放入0℃环境下进行第二次沉淀,沉淀出不溶于醇类有机溶剂的反应物,过滤得第二过滤液;
[0060]
c、重复步骤b三次,最后将滤液旋蒸得到产物,干燥处理后,密封保存。
[0061]
实施例2对苯二甲酸乙二醇酯的制备
[0062]
分别称取1mol的pta和1.4mold的eg,同时将催化剂sb2o3投入反应釜中,酯化温度为240℃,酯化反应时间为3h,生成对苯二甲酸乙二醇酯;
[0063][0064]
实施例3~6聚酯酰胺的制备
[0065]
将实施例1制得的pa56盐衍生物与实施例2制得的对苯二甲酸乙二醇酯(bhet)按照表1中的添加量加入反应釜内,进行缩聚反应,缩聚阶段反应温度见表1,缩聚反应的时间为3.5h,真空度控制在50pa以下,反应结束后出料、切粒、干燥。预结晶方式:在135℃预结晶1.5h后,利用真空转鼓在145℃下干燥22小时,得干燥切片,铝塑袋密封保存。
[0066]
表1
[0067]
样品编号bhet/molpa56盐衍生物/mol缩聚阶段反应温度实施例31.10.05270℃实施例41.40.10260℃实施例51.30.15255℃实施例61.20.20250℃
[0068]
鉴定数据:采用bruker 600型核磁共振波谱仪。对实施例3~6样品的结构进行表征,内部基准物为四甲基硅烷(tms)。称取5~10mg样品放入样品管内,用氘代三氟乙酸(cf3cood)试剂溶解,样品完全溶解后,将样品管放入探头中进行测试,1h-nmr扫描次数为16,测试结果见图1、图2。
[0069]
采用nicolet 6700型傅里叶红外光谱仪,对实施例3~6样品中的特征官能团进行分析,扫描光谱范围为4000~400cm-1
,分辨率为4cm-1
,扫描次数为10。经检测,在727cm-1
处
出现苯环上h的弯曲振动峰;1718cm-1
处出现-c=o的伸缩振动峰;873cm-1
处出现苯环上2个相邻h的伸缩振动峰;1095cm-1
和1244cm-1
处分别出现-c-o-c和-coo-的伸缩振动峰,以上均为聚酯上的特征吸收峰。而1656cm-1
处出现酰胺吸收带(c=o的伸缩振动),1534cm-1
处出现酰胺吸收带(n-h的弯曲振动)。
[0070]
根据以上检测结果推知得到实施例3~6(图1中的5#~8#分别对应实施例3~6)的聚酯酰胺的结构式如下:
[0071][0072]
实施例7和8聚酯酰胺的制备
[0073]
实施例7
[0074]
与实施例5的不同之处仅在于制备方法:缩聚反应的温度为235℃。与实施例5相比,在该温度下反应,相同时间下体系大部分聚合物的聚合粘度低,得到的聚酯酰胺的强度较低,纺丝容易断丝,不适合用于大规模纺丝。
[0075]
实施例8
[0076]
与实施例5的不同之处仅在于制备方法:缩聚反应的温度为280℃。由于缩聚温度较高,聚合物分子产生了部分交联。
[0077]
实施例9聚酯酰胺纤维的制备
[0078]
将实施例3~8以及对比例1中保存的产品转移至纺丝机,采用螺杆挤出机加热原料至熔融态;通过计量泵准确计量后,从喷丝板的喷丝孔挤出,形成预取向丝,即,聚酯酰胺poy纤维。其中,纺丝螺杆挤压机采用五区加热,一区温度250℃、二区温度272℃、三区温度280℃、四区温度282℃、五区温度278℃、纺丝箱温度278℃,纺丝组件压力12mpa,纺丝速度3500m/min。再将得到的聚酯酰胺poy纤维在130℃下进行牵伸热定型,牵伸倍数为1.5倍,得到聚酯酰胺fdy纤维。
[0079]
对比例1pet的制备
[0080]
将实施例2制得的bhet进行缩聚反应,缩聚阶段反应温度为275℃,真空度控制在50pa以下,反应结束后出料、切粒、干燥,即可得到pet。预结晶方式同上述实施例。
[0081]
鉴定数据:测试方式同上述实施例3~6,根据图1和图2中图谱推知得到对比例1中pet(图1中的0#)的结构式与现有的pet结构式一致。
[0082]
效果实施例1
[0083]
将实施例3~6和对比例1中的聚酯酰胺与pet进行压膜,压膜温度为220~260℃,膜厚约1mm,将压膜片进行如下测试:
[0084]
(1)x射线衍射(xrd)测试
[0085]
采用d/max-2550vb+/pc*型x射线衍射仪,针对高聚物内部存在密度不匀现象,研究高聚物的结晶性能。实验在室温进行,先将样品在80℃真空干燥烘箱中真空干燥2h,辐射靶源为cu靶,波长为15.4nm,电压为40kv,扫描范围为5
°
~60
°
,测试的waxd数据如表2所示。
[0086]
表2
[0087]
样品编号结晶度/%对比例144.20
实施例333.97实施例432.73实施例528.97实施例626.88
[0088]
由表2可知,本技术实施例3~6的聚酯酰胺的结晶度在24.09%~33.97%之间,随着改性单体添加量的增加,聚酯酰胺的结晶度逐渐降低,非晶区增加,有利于分散染料染色。
[0089]
(2)差示扫描量热(dsc)测试
[0090]
采用dsc250差示扫描量热仪。先以20℃/min的速率从室温升温到300℃,再以20℃/min的速率冷却至0℃,然后以10℃/min的速率从0℃升温到300℃,得到dsc曲线,氮气保护,测试结果如表3所示。
[0091]
表3
[0092]
样品编号tg/℃对比例177.8实施例364.5实施例459.1实施例548.4实施例646.9
[0093]
由表3可知,本技术实施例3~6的聚酯酰胺的tg低于对比例1的tg;而tg的高低很大程度上受分子链的柔顺性影响,分子链的柔顺性越好,tg越低,越有利于低温染色。
[0094]
(3)热重分析(tga)测试
[0095]
采用tg4000热失重仪。称取5~10mg样品放入坩埚中,升温速率为20℃/min,升温范围为30~700℃,氮气保护,测试结果如表4所示。
[0096]
表4
[0097][0098]
在整个受热过程中,由表4可知,本技术实施例3~6的聚酯酰胺起始降解温度在386~404℃之间,最大失重速率温度424~435℃之间,远高于纺丝温度(200℃左右)。
[0099]
(4)特性粘度测试
[0100]
按照国标gb/t 14190-2008纤维级聚酯切片试验方法进行测试,所用乌式粘度计的毛细管直径在0.8mm,测试时将苯酚与四氯乙烷按质量比1:1混合,将样品溶解其中配置
成浓度为0.005g/ml的溶液。将溶剂以及各待测溶液放在25
±
0.1℃的恒温槽中恒温10min,观察并记录溶剂和各待测溶液流过两条刻度线所用时间,每个样品测试三次,前后两次时间误差在0.2s之内,然后取三次测试的平均值,最后按公式(1-1)和(1-2)进行特性粘度[η]计算,并根据公式(1-3)计算出相应高聚物的粘均分子量m
η
。
[0101][0102][0103]
式中:[η]为特性粘度,单位dl/g;ηr为相对粘度;η0为纯溶剂的粘度;η
sp
为增比粘度;t0为溶剂流出时间,单位s;t为溶液流出时间,单位s;c为溶剂浓度,单位g/ml。
[0104]
[η]=km
ηα
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(1-3)
[0105]
式中:m
η
为粘均分子量;比例常数k为2.1
×
10-4
;扩张因子α为0.82。
[0106]
表5
[0107][0108]
由表5可知,本技术实施例3~6制得的聚酯酰胺的特性粘度在0.65~0.78dl/g之间,对应的粘均分子量在18070~22570之间,能够达到聚酯类纤维的可纺级别。
[0109]
(5)静态接触角测试
[0110]
采用kino sl200b动/静态接触角测角仪(美国科诺工业有限公司)。用双面胶将样品粘贴在载玻片的中间,测试面朝上,试样尺寸为40mm
×
5mm,测试样品的静态接触角,每个样品测试5个位点取平均值。
[0111]
测试结果如下:
[0112]
对比例1的pet的静态接触角为91.2℃,实施例3~6样品的静态接触角分别为82.3℃、77.4℃、73.6℃和68.1℃,和纯pet静态接触角相比大幅度下降,且随着改性单体添加量的增加而逐渐降低,聚合物从疏水变为亲水。
[0113]
效果实施例2
[0114]
对实施例9中制得的聚酯酰胺纤维(采用实施例3~6和实施例8的聚酯酰胺制备)进行上染率测试、染色m率、条干不均率测试,测试结果如表6所示。
[0115]
(1)上染率的测定方法为:用分光光度计测试染色前后染液浓度的变化。上染率(%)=(a0-at)/a0*100%;其中,a0为处理前的染料特征吸收峰的吸光度值,at为处理时间t时的染料吸光度。
[0116]
由表6可知,采用实施例3~6聚酯酰胺制得的聚酯酰胺纤维在100℃的上染率均在
80%以上,甚至可达到99%以上,远远高于对比例1中的上染率;在130℃时的上染率也均在98%以上,仍然高于对比例1的上染率,也就是说本技术的聚酯酰胺纤维不仅可以保证高温条件下的上染率,而且还能明显提升较低温度下的上染率。
[0117]
(2)染色m率
[0118]
采用染色均匀度方法(astmz7667-1999)测定上述制备方法制备得到聚酰胺纤维的染色均匀度,并采用5级判色标准方法测定m率。m率=(染色均匀度≥4.5级的纤维数量)/所有染色纤维总数量)
×
100%。
[0119]
由表6可知,采用实施例3~6聚酯酰胺制得的聚酯酰胺纤维的染色m率均在99%以上,染色均匀度较高。
[0120]
表6
[0121]
样品编号100℃分散染料上染率/%130℃分散染料上染率/%染色m率/%对比例118.796.298.5实施例383.298.899.0实施例485.699.199.2实施例598.799.699.7实施例699.299.499.0
[0122]
(3)条干不均率测试。
[0123]
条干不均率:按gb/t 14346-93方法进行测定。测试结果如表7所示。
[0124]
由表7可知,本技术实施例5和8聚酯酰胺制得的聚酯酰胺纤维的条干不均率均在2%以下,甚至可以达到1.2%左右,说明聚酯酰胺纤维的结构均匀较高,纺丝效果好。
[0125]
表7
[0126][0127]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1