一种神经递质再摄取抑制剂的晶型及其制备方法和用途
1.本发明属于药物化学领域,具体涉及一种五羟色胺及去甲肾上腺素(5-ht/ne)双重再摄取抑制剂的晶型及其制备方法和用途。
背景技术:
2.抑郁症又称抑郁障碍,是一种以情绪低落、思维迟缓、意志活动减退为主要特征的情感性精神障碍,具有高复发、高患病、高医疗成本、高致残的特点,临床上主要表现为认知功能损害和躯体症状。多数患者存在厌世乃至轻生的念头,影响患者的生命质量,给家庭和社会造成沉重的心理压力和经济负担。[顾然,等,“抑郁症发病机制研究进展”,《养生保健指南》,2019,33:262;米智华,等,“抑郁症的发病机制及针刺治疗研究进展”,《实用临床医药杂志》,2019,23(8):123-127。]
[0003]
抑郁症病因复杂,目前尚未阐明,普遍认为可能与遗传因素、生物学因素、社会心理等多种因素相关。关于抑郁症的发病机制,目前主要有5-ht假说、去甲肾上腺素(ne)假说、多巴胺假说、乙酰胆碱假说、氧化应激与神经炎症等,其中5-ht假说已被广泛认可和接受,ne转运体也已经成为重度抑郁症的一种靶向治疗途径。研究表明,增加血5-ht水平的药物,如选择性5-ht再摄取抑制剂,可有效治疗抑郁和焦虑,采用mri和pet扫描重度抑郁症患者丘脑及其亚区,发现在重度抑郁症患者丘脑及其亚区的去甲肾上腺素转运体可用性增高。[米智华,等,“抑郁症的发病机制及针刺治疗研究进展”,《实用临床医药杂志》,2019,23(8):123-127。]
[0004]
焦虑障碍(anxiety disorders)又称焦虑症,是一组以持续性焦虑、紧张为主的神经症。焦虑障碍包括惊恐障碍、混合性焦虑、抑郁障碍、广泛性焦虑症、恐慌症、广场恐怖症、强迫症、社交恐怖症和创伤后精神紧张性障碍等,其有两种主要的临床形式:惊恐障碍和广泛性焦虑。焦虑障碍药物治疗主要用以减轻焦虑、紧张、恐惧、稳定情绪,治疗药物包括苯二氮卓类、5-ht 1a受体激动剂类、咪唑吡啶类、吡咯环酮类、吡唑嘧啶类和β-肾上腺素受体阻断剂等。
[0005]
疼痛是一种因实际的或潜在的组织损伤而产生的痛苦感觉,常伴有不愉快的情绪或心血管和呼吸方面的变化。根据持续时间的不同,疼痛可分为急性疼痛和慢性疼痛。急性疼痛多是由组织创伤引起的伤害性疼痛,例如急性炎性疼痛、肌纤维痛等,而慢性疼痛是以神经病理性疼痛为主的疾病,例如慢性炎性疼痛、神经源性疼痛等。疼痛在临床发病率高,给患者带来极大痛苦;此外,疼痛与抑郁症等精神疾病有很高的共患病比例,而多数抑郁症患者也伴发慢性疼痛。
[0006]
传统镇痛药物主要包括阿片类药物和非甾体抗炎药。阿片类镇痛药镇痛作用强,但长期使用易导致耐受性、依赖性和成瘾性,并有呼吸抑制、中枢镇静等不良反应,目前用于急性锐痛和癌性剧痛等。非甾体抗炎药仅发挥中等程度镇痛作用,适用于轻度和中度的慢性钝痛,但对直接刺激感觉末梢引起的锐痛无效,此外还具有消化道出血和心脏毒性等不良反应。
[0007]
化合物i是一种五羟色胺及去甲肾上腺素(5-ht/ne)双重再摄取抑制剂,中国专利cn102850335a中公开了式i及其异构体、制备方法及其作为抗抑郁药的用途,性状为白色粉末状固体,熔点为132-133℃。化合物i对5-ht转运蛋白和ne转运蛋白的亲和力与度洛西汀相当,抗抑郁活性优于度洛西汀。另外,中国专利cn109758454a和cn109758455a中也分别公开了游离形式的式i化合物具有抗焦虑作用和镇痛作用,且效果均优于度洛西汀。
[0008][0009]
在合成和初步纯化后,中国专利cn102850335a并未对药品在后续开发中可能存在的缺陷及如何克服这些缺陷进行探索,也没有对是否存在多晶型进行研究,由于药物晶型直接关系到药物的质量及疗效,不同的晶型会不同程度地影响药物稳定性、生物利用度和安全性,因此,发现性质优良的晶型对后续药品研发十分关键。
[0010]
发明人按中国专利cn102850335a中的方法制备了化合物i,发现所得产品稳定性较差,影响因素试验表明产品中单杂和总杂增长较快,不能满足药物的存储和安全性需要。为了解决这些问题,发明人对化合物i的晶型进行了大量试验研究,终于得到了一种纯度高、晶体颗粒外观形态良好且规则、无引湿性、稳定性好、流动性好的化合物i晶型a,适宜作为原料药品存储和使用,克服了现有技术存在的缺陷。
技术实现要素:
[0011]
发明人在cn102850335a的基础上对化合物i的晶型进行系统研究发现,在多种晶体研究方法和不同试验条件下不能得到结晶固体,仅能获得油状物或胶状物。经多次尝试之后,仅获得本发明晶型a,且未发现其他晶型。
[0012]
一方面,本发明提供了一种化合物i的晶型a,其熔点为148~154℃。
[0013]
本发明的一些方案中,所述晶型a,其差示扫描量热曲线在150.14
±
3℃有一个吸热峰的起始点。
[0014]
本发明的一些方案中,所述晶型a,其具有基本上如图1所示的dsc图谱。
[0015]
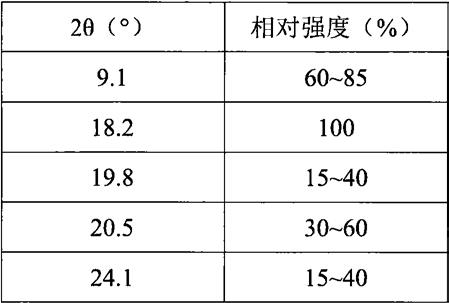
本发明的一些方案中,所述晶型a,使用cu-kα辐射,以2θ角度(
°
)表示的粉末x-射线衍射图谱在以下位置有特征衍射峰:9.1
±
0.2
°
,18.2
±
0.2
°
,19.8
±
0.2
°
,20.5
±
0.2
°
,25.4
±
0.2
°
。
[0016]
优选地,上述特征峰的相对强度为:
[0017]
2θ(
°
)相对强度(%)9.160~8518.210019.815~4020.530~6025.430~60
[0018]
本发明的一些方案中,所述晶型a,其特征在于,使用cu-kα辐射,以2θ角度(
°
)表示的粉末x-射线衍射图谱在以下位置有特征衍射峰:9.1
±
0.2
°
,18.2
±
0.2
°
,19.8
±
0.2
°
,
20.5
±
0.2
°
,24.1
±
0.2
°
,25.4
±
0.2
°
,30.0
±
0.2
°
。
[0019]
优选地,上述特征峰的相对强度为:
[0020][0021][0022]
本发明的一些方案中,所述晶型a,其特征在于,使用cu-kα辐射,以2θ角度(
°
)表示的粉末x-射线衍射图谱在以下位置有特征衍射峰:9.1
±
0.2
°
,16.4
±
0.2
°
,18.2
±
0.2
°
,19.8
±
0.2
°
,20.5
±
0.2
°
,24.1
±
0.2
°
,25.4
±
0.2
°
,27.4
±
0.2
°
,30.0
±
0.2
°
。
[0023]
优选地,上述特征峰的相对强度为:
[0024]
2θ(
°
)相对强度(%)9.160~8516.410~3518.210019.815~4020.530~6024.115~4025.430~6027.410~3530.015~45
[0025]
本发明的一些方案中,所述晶型a,其特征在于,使用cu-kα辐射,以2θ角度(
°
)表示的粉末x-射线衍射图谱在以下位置有特征衍射峰:9.1
±
0.2
°
,16.4
±
0.2
°
,18.2
±
0.2
°
,19.8
±
0.2
°
,20.0
±
0.2
°
,20.5
±
0.2
°
,20.9
±
0.2
°
,24.1
±
0.2
°
,25.4
±
0.2
°
,27.4
±
0.2
°
,30.0
±
0.2
°
。
[0026]
优选地,上述特征峰的相对强度为:
[0027][0028][0029]
本发明的一些方案中,所述晶型a,使用cu-kα辐射,其粉末x-射线衍射图谱基本上如图2所示。
[0030]
本发明的一些方案中,所述晶型a,其xrpd图谱解析数据如下表所示:
[0031][0032]
本发明的一些方案中,所述晶型a,其结晶颗粒具有长方体棒状或片状的晶体形状。
[0033]
本发明的一些方案中,所述晶型a,其结晶颗粒具有基本上如图3所示的晶体形状。
[0034]
另一方面,本发明还提供上述晶型a的制备方法,该方法包括:将化合物i的粗品加入溶剂1和溶剂2组成的混合溶剂中,加热回流至全部溶解,缓慢降温析晶,分离得晶型a。
[0035]
本发明的一些方案中,上述制备方法,其中,所述溶剂1为roh,溶剂2选自rcor1、rcn或rcln,其中r和r1各自独立地选自c
1-c4直链或支链烷基,n为1-4;优选地,溶剂1选自异丙醇、乙醇或甲醇,溶剂2选自丙酮、乙腈或二氯甲烷;进一步优选地,溶剂1为异丙醇,溶剂2为丙酮。
[0036]
本发明的一些方案中,上述制备方法,其中,所述溶剂1和溶剂2的体积比为1~5∶1~100,优选为1~3∶1~50,更进一步优选为1~3∶1~20,更进一步优选为1~3∶1~10。
[0037]
本发明的一些方案中,上述制备方法,其中,化合物i粗品与溶剂1的质量体积比为1g∶0.5~30ml,优选为1g∶0.5~20ml,进一步优选为1g∶0.5~15ml,更进一步优选为1g∶1~10ml。
[0038]
本发明的一些方案中,上述制备方法,其中,所述分离步骤包括采用过滤、离心等
适宜的方法将所得晶型a从结晶液中分离出来。
[0039]
本发明的一些方案中,上述制备方法,其中,所述加热的温度为20℃~回流温度,优选为50℃~回流温度。
[0040]
本发明的一些方案中,上述制备方法,其中,所述降温析晶温度为-30℃~30℃,优选为-20℃~25℃,进一步优选为-5℃~20℃,更进一步优选为0℃~10℃;或者,所述降温析晶温度进一步优选为0~30℃,更进一步优选10~30℃,更进一步优选20~30℃。
[0041]
本发明的一些方案中,上述制备方法,从去除产品中游离溶剂考虑,在分离步骤后,还包括干燥步骤,干燥方法可采用任何适宜的已知方法,优选为减压(真空)干燥。具体的干燥条件是,例如,温度优选40~60℃,更优选为40~50℃;干燥时间优选为4~20h,更优选为5~8h。无论采用何种干燥手段,都以所得产品中溶剂残留量符合质量标准为宜。
[0042]
本发明中所述的化合物i粗品或化合物i,采用cn102850335a中公开的已知方法进行制备,也可使用其它现有技术中公开的任何已知方法进行制备。
[0043]
本发明另一方面还提供一种药物组合物,包含上述晶型a或按上述制备方法制备的晶型a及药学上可接受的载体,任选地,所述药物组合物还可包含其他治疗组分。所述其它治疗组分是指预防和/或治疗抑郁症、焦虑障碍或疼痛的其它活性成分或药物。优选的,所述其他治疗组分能与化合物i产生协同作用。
[0044]
上述药物组合物制成临床接受的制剂,例如口服制剂、注射制剂、局部给药制剂、外用制剂等,优选口服制剂。所述口服制剂优选固体制剂,如片剂、胶囊、颗粒剂等。这些制剂可采用本领域一般技术人员公知的相应辅料,采用相应公知的药物制剂的制备技术制得。
[0045]
另一方面,本发明还提供了上述晶型a、或按上述制备方法制备的晶型a或包含晶型a的药物组合物在制备5-ht/ne双重再摄取抑制剂相关药物中的应用。
[0046]
本发明的一些方案中,上述5-ht/ne双重再摄取抑制剂相关药物是用于预防和/或治疗抑郁症、焦虑障碍、疼痛的药物;其中,所述焦虑障碍选自惊恐障碍、广泛性焦虑症、恐慌症、广场恐怖症、强迫症、社交恐怖症或创伤后精神紧张性障碍,所述疼痛选自急性炎性疼痛、慢性炎性疼痛、神经源性疼痛或肌纤维痛。
[0047]
另一方面,本发明还涉及上述晶型a、或按上述制备方法制备的晶型a或包含晶型a的药物组合物,其用于预防和/或治疗5-ht/ne相关病症,所述5-ht/ne相关病症为抑郁症、焦虑障碍或疼痛;其中,所述焦虑障碍选自惊恐障碍、广泛性焦虑症、恐慌症、广场恐怖症、强迫症、社交恐怖症或创伤后精神紧张性障碍,所述疼痛选自急性炎性疼痛、慢性炎性疼痛、神经源性疼痛或肌纤维痛。
[0048]
另一方面,本发明还涉及一种治疗患者的方法,通过向患者施用上述晶型a、或按上述制备方法制备的晶型a或包含晶型a的药物组合物,所述患者的病症为5-ht/ne相关病症。
[0049]
本发明的一些方案中,所述5-ht/ne相关病症为抑郁症、焦虑障碍或疼痛;其中,所述焦虑障碍选自惊恐障碍、广泛性焦虑症、恐慌症、广场恐怖症、强迫症、社交恐怖症或创伤后精神紧张性障碍,所述疼痛选自急性炎性疼痛、慢性炎性疼痛、神经源性疼痛或肌纤维痛。
[0050]
上述的“患者”包括动物界的所有成员,包括但不限于,哺乳动物(例如,小鼠、大
鼠、猫、猴子、狗等)和人。
[0051]
术语“基本上如附图所示”是指基本上纯净的某种晶型其粉末x-射线衍射图谱或dsc图谱或结晶颗粒的晶体形状图中至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或至少95%,或至少96%,或至少97%,或至少98%,或至少99%的峰出现在所给出图谱中。进一步的,当产品中某种晶型的含量逐渐降低时,其粉末x-射线衍射图谱中的一些归属于该晶型的衍射峰可能会由于仪器的检测灵敏度的因素而变少。
[0052]
定义和说明
[0053]
除非另有说明,本文所用的下列术语和短语旨在含有下列含义。一个特定的短语或术语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文出现商品名时,旨在指代其对应的商品其活性成分。
[0054]
本发明的中间体化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。
[0055]
本发明具体实施方式的化学反应是在合适的溶剂中完成的,所述的溶剂须适合于本发明的化学变化及其所需的试剂和物料。为了获得本发明的化合物,有时需要本领域技术人员在已有实施方式的基础上对合成步骤或者反应流程进行修改或选择。
[0056]
下面会通过实施例具体描述本发明,无需进一步纯化即可使用。
[0057]
本发明所使用的溶剂可经市售获得。
[0058]
本发明采用下述缩略词:
[0059]
dmac:n,n-二甲基乙酰胺;
[0060]
dmso:二甲基亚砜;
[0061]
etoac:乙酸乙酯;
[0062]
ipac:醋酸异丙酯;
[0063]
naoh:氢氧化钠;
[0064]
nmp:n-甲基吡咯烷酮;
[0065]
mek:甲基乙基酮;
[0066]
mibk:甲基异丁酮;
[0067]
thf:四氢呋喃。
[0068]
技术效果
[0069]
发明人对化合物i的晶型进行了系统研究,在cn102850335a中公开的白色粉末状固体(熔点为132-133℃)的基础上,未发现除本发明晶型a以外的其他晶型。在多种晶体研究方法和不同试验条件下甚至不能得到固体,仅能获得油状物或胶状物。
[0070]
本发明的晶型a的有益效果在于:
[0071]
(1)本发明的晶型a外观性状良好,为结晶状粉末,结晶颗粒微观形态分布均匀、有规则,具有长方体棒状或片状的晶体形状。
[0072]
(2)本发明的晶型a纯度高,且具有优良的稳定性。将本发明所得的晶型a置于高温、高湿和强光条件下,被考察样品的最大单杂、总杂和含量均无明显变化,晶型未改变,更适宜作为原料药物储存和使用。
[0073]
(3)本发明的晶型a粒度均匀,流动性和可压性好,适宜进行药物制剂开发。
[0074]
(4)本发明的晶型a的制备工艺操作简单,更适于在工业上使用。
附图说明
[0075]
图1:实施例1所得晶型a的dsc谱图。
[0076]
图2:实施例1所得晶型a的粉末x-射线衍射图。
[0077]
图3:实施例1所得晶型a的结晶颗粒显微镜图。
具体实施方式
[0078]
1、粉末x-射线衍射(x-ray powder diffractometer,xrpd)
[0079]
仪器型号:布鲁克d2 phaser x射线衍射仪
[0080]
样品用量:100mg
[0081]
靶:cu(30kv,10ma)
[0082]
阶跃角:0.02
°
[0083]
扫描范围:3.0~40.0
°
[0084]
扫描速度:0.02
°
/0.50s
[0085]
发射狭缝:0.2mm
[0086]
空气散射挡板:1mm
[0087]
接收器狭缝:8mm
[0088]
2、熔点
[0089]
熔点仪:jh70全自动视频熔点仪
[0090]
样品用量:10mg
[0091]
初始设定温度:80℃
[0092]
升温终点温度:160℃
[0093]
升温速率:10℃/min
[0094]
3、差热分析(differential scanning calorimeter,dsc)
[0095]
仪器型号:dsc-tga6差示扫描量热仪
[0096]
测试方法:取样品(2mg)置于dsc铝锅内进行测试,在50ml/min n2条件下,以10℃/min的升温速率,加热样品从80℃到170℃。
[0097]
4、引湿性
[0098]
采用中国药典(2015年版,第四部)中的方法进行测定,具体试验方法如下:
[0099]
1)取干燥的具塞玻璃称量瓶(外径为50mm,高为15mm),于试验前一天置于适宜的25℃
±
1℃恒温干燥箱(下部放置氯化铵或硫酸铵饱和溶液)或人工气候箱(设定温度为25℃
±
1℃,相对湿度为80%
±
2%)内,精密称定重量(m1);
[0100]
2)取供试品适量,平铺于上述称量瓶中,供试品厚度一般约为1mm,精密称定重量(m2);
[0101]
3)将称量瓶敞口,并于瓶盖同置于上述恒温恒湿条件下24h;
[0102]
4)盖好称量瓶盖子,精密称定重量(m3);
[0103]
增重百分率=(m
3-m2)/(m
2-m1)
×
100%;
[0104]
5)引湿性特征描述与引湿性增重的界定:
[0105][0106][0107]
5、结晶颗粒外观检测
[0108]
设备名称:dmi-3000b倒置荧光显微镜
[0109]
捕抓分辨率:3072
×
2048
[0110]
设置比例尺:1
[0111]
选择标尺:1x
[0112]
设置线条宽度:1
[0113]
6、溶解度实验
[0114]
设备名称:mettler s210-s型ph计、agilent1200型高效液相色谱仪、sha-b型水浴恒温振荡器
[0115]
检测溶液:纯化水,另外配制磷酸氢二钠-柠檬酸缓冲液(ph 4.5)和磷酸氢二钠-柠檬酸缓冲液(ph 6.8)
[0116]
色谱柱:用十八烷基硅烷键合硅胶为填充剂
[0117]
流动相:以0.025mol/l磷酸二氢钾溶液(含0.1%三乙胺,用磷酸调ph6.0)为流动相a,以甲醇为流动相b
[0118]
洗脱梯度:
[0119][0120]
流速:1.0ml/min
[0121]
检测波长:230nm
[0122]
进样体积:20μl
[0123]
供试品溶液制备:取纯化水、ph4.5缓冲盐溶液及ph6.8缓冲盐溶液各20ml,分别置4支洁净试管中。加供试品过量,置水浴振荡器中于37.0℃下振摇24小时,得不同溶剂的供试品饱和溶液。稀释至适宜浓度,作为供试品溶液。
[0124]
对照品溶液制备:取对照品约25mg,精密称定。置25ml量瓶中,加适量水溶解,并稀释至刻度,摇匀。
[0125]
测定方法:取供试品溶液与对照品溶液各20μl,注入高效液相色谱仪,记录色谱
图。以外标法计算供试品在不同介质中的溶解度。
[0126]
7、粒度检测
[0127]
设备名称:马尔文激光粒度测定仪(马尔文2000)
[0128]
测定方法:干法
[0129]
样品用量:约2.0g
[0130]
遮光度:1%
[0131]
折射率:1.52
[0132]
测定气压:2.0bar
[0133]
测量时间:15s
[0134]
背景测定时间:10s
[0135]
8、粉体学参数检测
[0136]
设备名称:bt-1001智能粉体特性测试仪
[0137]
休止角测试参数:样品量:约50g;测试次数:3次;进料速度:4:进料时间:200s
[0138]
松装密度测试参数:样品量:约50g;进料速度:4,进料时间:300s
[0139]
振实密度测试参数:样品量:约50g;振实频率:100,,震动次数:500
[0140]
卡尔系数(%)=(振实密度-松装密度)/振实密度
×
100%
[0141]
豪斯纳比指数=振实密度/松装密度。
[0142]
以下将参照附图,对本发明的优选实施例进行详细描述。所举实施例是为了更好地对本发明的内容进行说明,但并不是本发明的内容仅限于所举实施例。本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
[0143]
制备例1化合物i的制备
[0144]
参照cn102850335a中公开的方法进行化合物i的制备,得白色粉末状固体75.5g,熔点:132~133℃。
[0145]
实施例1化合物i的晶型a的制备
[0146]
将制备例1样品63.5g加入到异丙醇/丙酮(1400ml,v∶v=1.1∶1)的混合溶剂中,加热回流至固体全部溶解,继续回流30min,趁热过滤,滤液再次回流10min,缓慢降温至25℃
±
5℃,析晶12h,过滤,45℃真空干燥12h,得白色结晶状粉末(47.6g,收率74.96%),纯度99.66%,熔点151.1-152.8℃。取样品进行dsc测试,其dsc曲线在150.14℃有一个吸热峰的起始点,见附图1。取样品进行x-射线粉末衍射,显示为晶型a,结果见表1,谱图见附图2。结晶颗粒具有长方体棒状或片状的晶体形状,见附图3。
[0147]
表1实施例1所得的晶型a粉末-x射线衍射特征峰数据
[0148][0149]
实施例2-5化合物i的晶型a的制备
[0150]
参照实施例1制备方法,改变混合溶剂组成及比例。具体实验条件及结果如下:
[0151]
表2实施例2-5实验结果
[0152][0153]
实施例2-5所得产品性状及晶型xrpd图与实施例1基本相同。
[0154]
实验例1影响因素实验
[0155]
取实施例1晶型a样品适量,置于药用低密度聚乙烯袋中,分别在高温(60℃
±
2℃)、高湿(92.5%rh)、强光(4500lx)条件下放置10天后,与0天数据比较,实验结果见表3。
[0156]
表3实施例1晶型a的影响因素实验结果
[0157][0158]
将制备例1样品置于西林瓶中密封,在高温(60℃)条件下放置8天后,与0天数据比较,实验结果见表4。
[0159]
表4制备例1样品在高温试验中的实验数据结果
[0160][0161]
结论:实施例1晶型a在高温、高湿和强光下均较稳定,含量未发生明显改变,最大单杂、总杂也无明显变化趋势,且经检测,晶型亦未发生改变,即晶型a质量稳定,适宜作为原料药进行存贮。而制备例1样品在高温条件下放置8天后,最大单杂、总杂含量明显增加,稳定性较差,不符合药典对药品杂质限度的一般要求,不适合作为原料药。
[0162]
实验例2引湿性实验
[0163]
取实施例1晶型a样品适量,进行引湿性实验,结果见表5。
[0164]
表5实施例1晶型a样品的引湿性结果
[0165]
产品实施例1晶型引湿性/%0.02不变
[0166]
结论:实施例1所得晶型a无引湿性,适宜作为原料药存储。
[0167]
实验例3溶解度实验
[0168]
取实施例1晶型a样品适量,进行溶解度实验,结果见表6。
[0169]
表6实施例1晶型a样品在不同检测溶液中的溶解度结果
[0170]
检测溶液实施例1溶解度(g/100ml)晶型ph4.5缓冲液7.849不变ph6.8缓冲液6.305不变纯化水5.045不变
[0171]
结论:实施例1所得晶型a在不同溶剂体系内,均具有良好的溶解度,且晶型维持稳定,即在生理条件下,不会出现在服药后晶型转变的现象。
[0172]
实验例4粉体学性能检测实验
[0173]
取实施例1晶型a样品适量,进行粉体学性能(粒度、休止角、松装密度和振实密度)的检测,结果见表7、表8和表9。
[0174]
表7实施例1晶型a样品的粒度测试结果
[0175][0176]
表8实施例1晶型a样品的休止角测试结果
[0177][0178][0179]
注:休止角31-35
°
代表粉末流动性良好,休止角46-55
°
代表粉末流动性不大好(仪器附带操作说明书,或,王亮,等,“医药粉体流动性评价方法研究进展”,《中国粉体技术》,2016,22(5):28-34,第29页表1)。
[0180]
表9实施例1晶型a样品的松装密度和振实密度测试结果
[0181][0182]
注:卡尔系数16%-20%,豪斯纳比指数1.19-1.25,代表样品的流动性和可压缩性良好,卡尔系数≥38%,豪斯纳比指数>1.60,代表样品的流动性和可压缩性非常差(王亮,等,“医药粉体流动性评价方法研究进展”,《中国粉体技术》,2016,22(5):28-34,第29页表1)。
[0183]
结果:从表7的结果看,实施例1晶型a样品的粒度分布较均匀,三次取样得到的结果误差很小;从表8和表9的结果看,实施例1晶型a样品的流动性和可压缩性良好,在后续制剂工序中,良好的流动性将利于其与药用辅料混合均匀,良好的可压缩性将有利于制剂压片工序中控制片剂的脆碎度。
[0184]
对比例
[0185]
研发过程中,发明人发现,化合物i在多种晶体研究方法和不同试验条件下无法析出结晶状固体,仅能获得油状物或胶状物。示例性的方案包括但不仅限于以下对比例:
[0186]
对比例1缓慢降温法
[0187]
称量约20mg制备例1样品于3ml小瓶中,加入0.5ml下表所示的混合溶剂,50℃搅拌约2小时,趁热过滤得澄清溶液,缓慢降温至5℃,均无固体析出;转移至-20℃静置3天溶液仍保持澄清;室温挥发9天后未析出固体,转移至室温下抽真空2天,最终均得到油状物。
[0188][0189][0190]
对比例2反溶剂添加法
[0191]
将约20mg制备例1样品溶于0.65ml水中,过滤得澄清溶液,向溶液中加入3ml丙酮(acetone)后得到澄清溶液,转至5℃搅拌7天仍澄清,再转至-20℃静置6天后凝固,转移至室温挥发17天,得油状物。
[0192]
对比例3反溶剂添加法
[0193]
将约20mg制备例1样品溶于0.15ml dmso中得澄清溶液,向溶液中加入0.8ml醋酸异丙酯(ipac)后体系略浑浊,在室温下搅拌1.5小时后即观察到油状物。
[0194]
对比例4反反溶剂添加法
[0195]
称量约20mg制备例1样品于hplc小瓶中,加入溶剂1ml乙醇(etoh),过滤得澄清的样品溶液,在20ml小瓶中加入5ml反溶剂甲基乙基酮(mek),将样品溶液逐滴加入到反溶剂中并室温搅拌1天后未析出固体,转移至5℃搅拌1天后仍澄清,转移至室温挥发8天,得油状物(编号4-1)。
[0196]
编号溶剂反溶剂4-1乙醇(etoh)甲基乙基酮(mek)4-2乙腈(can)丙酮(acetone)4-3三氯甲烷(chcl3)正丁醇(n-butanol)
[0197]
按上表调整溶剂、反溶剂的种类(编号4-2、4-3),使用与对比例4-1相同的方法,试验过程中无固体析出,最终得油状物。
[0198]
对比例5缓慢挥发法
[0199]
称量约20mg制备例1样品于3ml小瓶中,加入2ml如下表所示的溶剂,过滤得澄清溶液,用封口膜封口后扎3个小孔,通风橱中静置室温挥发13天,得到产物。
[0200]
编号溶剂产物5-1三氯甲烷(chcl3)胶状物5-2乙醇油状物5-3乙腈/三氯甲烷(1∶1,v/v)油状物
[0201]
对比例6气液渗透法
[0202]
如下表所示,向约20mg制备例1样品中加入适量溶剂溶解,过滤至3ml小瓶中,在20ml小瓶中加入3ml反溶剂,将3ml小瓶敞口置于20ml小瓶内,将20ml小瓶密封。气液渗透7天无固体析出,转移至室温下挥发6天后无固体析出,转至室温抽真空2-8天,得到油状物。
[0203]
编号溶剂溶剂用量反溶剂6-1h2o0.65ml异丙醇(ipa)6-2h2o0.65ml乙腈
6-3h2o0.65ml丙酮6-4dmso0.2ml四氢呋喃(thf)6-5dmso0.2ml甲苯6-6dmf0.2ml乙腈
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1