一种室温下NBS参与制备S-取代基-半胱氨酸衍生物的方法
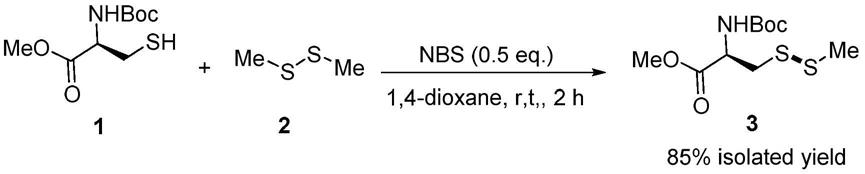
一种室温下nbs参与制备s
‑
取代基
‑
半胱氨酸衍生物的方法
技术领域
1.本发明涉及精细化工技术领域,尤其涉及一种室温下nbs参与制备s
‑
取代基
‑
半胱氨酸衍生物的方法。
背景技术:
2.s
‑
取代基半胱氨酸衍生物,作为一类重要的非对称二硫醚化合物,广泛存在于多种药物以及具有活性化合物中。因此,如何直接高效地合成该类化合物已经引起化学工作者的高度关注。在这些合成非对称二硫醚化合物的方法中,硫醇和对称二硫醚之间的直接交换反应是构建非对称二硫醚化合物最有效的方法之一。更重要的是硫醇与对称二硫醚直接的交换过程还被广泛应用于生物体系和自修复体系。最近,上海大学许斌等人使用氯化钯作为催化剂,dmso作为反应溶剂,在80℃实现了半胱氨酸衍生物和对称二硫醚之间的交换反应合成了s
‑
取代基
‑
半胱氨酸衍生物(sci论文:org.lett.2021,23,3167
‑
3172;中国专利:cn112047902)。尽管该方法可以合成s
‑
取代基
‑
半胱氨酸衍生物,但是需要使用昂贵的过渡金属钯催化剂,且反应温度较高,不符合绿色环保的特点。这些缺点限制了该策略在药物合成、生物体系和自修复体系中的应用。
技术实现要素:
3.针对上述背景技术,本发明的目的在于克服现有技术的不足并提供一种新的s
‑
取代基
‑
半胱氨酸衍生物的制备方法,采用nbs参与合成s
‑
取代基
‑
半胱氨酸衍生物,以对称的二硫醚化合物和半胱氨酸衍生物为原料,nbs(n
‑
溴代丁二酰亚胺)为添加剂,一锅法高效构建s
‑
取代基
‑
半胱氨酸衍生物。该方法不需要使用昂贵的钯金属催化剂,在室温下一步即可高效地合成s
‑
取代基
‑
半胱氨酸衍生物。需要说明的是,本发明说提供方法不仅仅局限于半胱氨酸衍生物,其它类型的硫醇包括芳基硫醇、杂芳基硫醇和烷基硫醇,也可以使用该方法合成相应的非对称的二硫醚化合物。
4.为了实现本发明的技术方案,所采用的技术方案为:
5.本发明提供一种室温下nbs参与制备s
‑
取代基
‑
半胱氨酸衍生物的方法,包括如下步骤:
6.将nbs、对称二硫醚和半胱氨酸衍生物于室温下反应,即得到所述s
‑
取代基
‑
半胱氨酸衍生物。
7.在本发明的技术方案中,所述反应在有机溶剂中进行,所述有机溶剂选自1,4
‑
二氧六环(1,4
‑
dioxane)、乙腈(mecn)、四氢呋喃(thf)、二氯乙烷(dce)、n,n
‑
二甲基甲酰胺(dmf)和二甲基亚砜(dmso)中的一种或任意几种混合使用,优选为1,4
‑
二氧六环。
8.在本发明的技术方案中,申请人发现当反应在1,4
‑
二氧六环中进行时,能够得到最高的产率。
9.在本发明的技术方案中,所述nbs为n
‑
溴代琥珀酰亚胺,cas号为128
‑
08
‑
5;所述对称二硫醚的结构通式为rssr,其中r选自甲基、乙基、丙基、异丙基、环己基、叔丁基、苯基、4
‑
氯苯基、4
‑
甲氧基苯基和2
‑
苯并噻吩基中的任一种;所述半胱氨酸衍生物的结构如式(i)所示:
[0010][0011]
式(i)中,r1选自
‑
h或
‑
boc(叔丁氧羰基),r2选自
‑
h或
‑
ch3。
[0012]
进一步地,所述室温下反应还包括分离纯化的操作。
[0013]
作为优选,所述nbs、对称二硫醚和半胱氨酸衍生物的摩尔比为0.2~1.0:1.0~3.0:1.0,优选为0.5:2.0:1.0。
[0014]
在某些具体的实施例中,所述nbs、对称二硫醚和半胱氨酸衍生物的摩尔比为0.2:3.0:1.0、0.5:3.0:1.0、0.5:2.0:1.0、0.7:2.0:1.0、1.0:1.0:1.0或它们之间的任意摩尔比。
[0015]
在本发明的技术方案中,申请人发现,nbs的摩尔比低于0.5时,产率降低,高于0.5时,产率略降低;而对称二硫醚的摩尔比低于2.0产率下降,高于2.0产率保持一致。
[0016]
作为优选,所述室温下反应的反应时间为1.0~6.0小时。
[0017]
作为优选,所述分离纯化包括浓缩反应液和柱层析分离。
[0018]
与现有技术相比,本发明取得了如下有益效果:本发明提供了一种s
‑
取代基
‑
半胱氨酸衍生物制备方法,用nbs参与制备半胱氨酸的不对称二硫醚衍生物,该方法不需要使用昂贵的钯金属催化剂,在室温下一锅法即可高效地合成s
‑
取代基
‑
半胱氨酸衍生物,产率最高可达85%。现有方法中,对称二硫醚的合成可通过nbs诱导硫醇发生反应制备,而本发明则发现了nbs可以进一步诱导对称二硫醚与硫醇反应生成不对称二硫醚。由于该方法具有在室温下且不需要使用昂贵的钯金属催化剂的优点,因此,该策略在药物合成、生物体系和自修复体系中具有潜在的应用性。
具体实施方式
[0019]
下述实施例仅仅是本发明的一部分实施例,而不是全部的实施例。因此,以下提供的本发明实施例中的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明的实施例,本领域技术人员在没有作出创造性劳动的前提下所获得的所有其他实施例,都属于本发明的保护范围。
[0020]
本发明下面结合实施例作进一步详述:
[0021]
本发明提供的制备方法的反应通式如下所示:
[0022][0023]
下述实施例中所用的试剂均为常规市售产品,所用方法无特意指出,均为本领域常规技术手段。其中半胱氨酸衍生物化合物1、化合物6、化合物8和化合物10均购买自毕得医药。下述实施例中所述室温的温度范围为20~30℃。
[0024]
实施例1:
[0025]
实施例1涉及的反应方程式如下:
[0026][0027]
n
‑
(叔丁氧羰基)
‑
s
‑
甲硫基
‑
l
‑
半胱氨酸甲酯的合成方法:在25ml的烧瓶中依次加入1,4
‑
二氧六环(5ml)、(叔丁氧羰基)
‑
l
‑
半胱氨酸甲酯1(117.7mg,0.5mmol)、二甲基二硫醚2(94.0mg,1.0mmol)和nbs(44.5mg,0.25mmol),室温并且剧烈搅拌反应2小时。反应结束后依次进行反应液浓缩和柱层析分离,即可获得n
‑
(叔丁氧羰基)
‑
s
‑
甲硫基
‑
l
‑
半胱氨酸甲酯3,收率85%(无色透明油,119.3mg)。其核磁共振氢谱为:1h nmr(300mhz,cdcl3)δ5.32(d,j=6.6hz,1h),4.57
–
4.54(m,1h),3.70(s,3h),3.15
–
3.02(m,2h),2.35(s,3h),1.38(s,9h).
13
c nmr(75mhz,cdcl3)δ171.36,155.08,80.20,52.84,52.56,40.22,28.29,23.08.lcms(esi,m/z):282.0[m+h]
+
。
[0028]
实施例2:
[0029][0030]
n
‑
(叔丁氧羰基)
‑
s
‑
苯硫基
‑
l
‑
半胱氨酸甲酯的合成方法:在25ml的烧瓶中依次加入1,4
‑
二氧六环(5ml)、(叔丁氧羰基)
‑
l
‑
半胱氨酸甲酯1(117.7mg,0.5mmol)、二苯基二硫醚4(218.3mg,1.0mmol)和nbs(44.5mg,0.25mmol),室温并且剧烈搅拌反应2小时。反应结束后依次进行反应液浓缩和柱层析分离,即可获得n
‑
(叔丁氧羰基)
‑
s
‑
苯硫基
‑
l
‑
半胱氨酸甲酯5,收率70%(无色透明油,120.2mg)。其核磁共振氢谱为:1h nmr(300mhz,cdcl3)δ7.52(d,j=7.5hz,2h),7.36
–
7.23(m,3h),5.32(d,j=7.2hz,1h),4.63
–
4.61(m,1h),3.74(s,3h),3.26
–
3.12(m,2h),1.44(s,9h).
13
c nmr(75mhz,cdcl3)δ171.12,155.03,136.62,129.16,128.35,127.43,80.26,52.85,52.64,40.89,28.31.lcms(esi,m/z):344.0[m+h]
+
。
[0031]
实施例3:
[0032][0033]
n
‑
(叔丁氧羰基)
‑
s
‑
苯硫基
‑
l
‑
半胱氨酸的合成方法:在25ml的烧瓶中依次加入1,4
‑
二氧六环(5ml)、(叔丁氧羰基)
‑
l
‑
半胱氨酸6(110.6mg,0.5mmol)、二苯基二硫醚4(218.3mg,1.0mmol)和nbs(44.5mg,0.25mmol),室温并且剧烈搅拌反应2小时。反应结束后依次进行反应液浓缩和柱层析分离,即可获得n
‑
(叔丁氧羰基)
‑
s
‑
苯硫基
‑
l
‑
半胱氨酸7,收率75%(白色固体,123.2mg)。其核磁共振氢谱为:1h nmr(300mhz,cdcl3)δ10.50(br,1h),
7.55(d,j=7.5hz,2h),7.38
–
7.25(m,3h),5.34(d,j=7.3hz,1h),4.67
–
4.54(m,1h),3.25
–
3.03(m,2h),1.47(s,9h).3c nmr(75mhz,cdcl3)δ175.32,155.40,136.46,129.19,128.60,127.55,80.74,52.85,40.35,28.31.lcms(esi,m/z):330.0[m+h]
+
。
[0034]
实施例4:
[0035][0036]
s
‑
甲硫基
‑
l
‑
半胱氨酸甲酯的合成方法:在25ml的烧瓶中依次加入1,4
‑
二氧六环(5ml)、l
‑
半胱氨酸甲酯8(67.6mg,0.5mmol)、二甲基二硫醚2(94.0mg,1.0mmol)和nbs(44.5mg,0.25mmol),室温并且剧烈搅拌反应2小时。反应结束后依次进行反应液浓缩和柱层析分离,即可获得s
‑
甲硫基
‑
l
‑
半胱氨酸甲酯9,收率80%(无色透明油,72.4mg)。其核磁共振氢谱为:1h nmr(300mhz,cdcl3)δ3.85
–
3.60(m,4h),3.07(dd,j=13.7,4.6hz,1h),2.84(dd,j=13.7,7.6hz,1h),2.36(s,3h).
13
c nmr(75mhz,cdcl3)δ174.23,53.45,52.35,42.60,22.97.lcms(esi,m/z):182.0[m+h]
+
。
[0037]
对比实施例1(不添加nbs):
[0038][0039]
在25ml的烧瓶中依次加入1,4
‑
二氧六环(5ml)、(叔丁氧羰基)
‑
l
‑
半胱氨酸甲酯1(0.5mmol)和二甲基二硫醚2(1.0mmol),室温并且剧烈搅拌反应2小时。反应结束后依次进行反应液浓缩和柱层析分离,不能分离到目标产物。
[0040]
对比实施例2(ncs为添加剂)
[0041][0042]
在25ml的烧瓶中依次加入1,4
‑
二氧六环(5ml)、(叔丁氧羰基)
‑
l
‑
半胱氨酸甲酯1(0.5mmol)、二甲基二硫醚2(1.0mmol)和ncs(n
‑
氯代丁二酰亚胺)(0.25mmol),室温并且剧烈搅拌反应2小时。反应结束后依次进行反应液浓缩和柱层析分离,不能分离到目标产物。本实施例中ncs指的是n
‑
氯代丁二酰亚胺。
[0043]
对比实施例3(二硫醚之间的交换反应)
[0044]
[0045]
在25ml的烧瓶中依次加入1,4
‑
二氧六环(5ml)、双(叔丁氧羰基)
‑
l
‑
半胱氨酸双甲酯1(0.5mmol)和二甲基二硫醚2(0.5mmol),室温并且剧烈搅拌反应2小时。反应结束后依次进行反应液浓缩和柱层析分离,不能分离到目标产物。
[0046]
通过上述以及实施例以及对比实施例可以看出:本发明以nbs参与制备半胱氨酸的不对称硫醚衍生物,其在常温下即可得到最高85%的产率,而采用ncs参与合成却无法得到半胱氨酸的不对称二硫醚,因此nbs是实现在室温下参与硫醇与对称二硫醚反应得到半胱氨酸的不对称二硫醚衍生物的关键试剂。
[0047]
以上所述仅为本发明的优选实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的等效变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1