一种三磷酸核苷酸盐的制备方法与流程
1.本发明涉及一种三磷酸核苷酸盐的制备方法。
背景技术:
2.随着核酸药物的发展,mrna被视为了一种可以用于药物制造的新选择。1990年,一段mrna被注射进入小鼠体内,并成功编码出了蛋白质。这段mrna则是通过一种名为体外转录的技术得到的。随后,一项1992年的研究发现注射抗利尿激素编码mrna可以成功诱导大鼠的下丘脑的神经活动。虽然mrna显示出很好的生物活性,但是受限于其自身的不稳定性,强免疫原性和体内递送困难,mrna远远无法应用于临床疾病治疗中。
3.修饰核苷酸的加入,可以降低mrna的自身免疫原性,提高mrna自身稳定性,进一步增强mrna在目标细胞内的表达时间和表达效率,使mrma可以真正用于制药领域。其中moderna和biontech分别采用1-甲基假尿苷(cn110511939a,cn104114572a,cn103974724a)和假尿苷(us10232055b2,us9597380b2,us9163213b2),极大的降低了mrna的免疫原性,并且提高了mrna在靶细胞内表达目的蛋白的时间和总量,为mrna制成covid-19疫苗并成功上市奠定了基础。
4.但是mrna合成用修饰三磷酸核苷酸制备工艺复杂,尤其是其纯化十分困难且产品稳定性差,导致修饰核苷酸商品售价高达数十万甚至上百元万元每克。现阶段高纯度的修饰三磷酸核苷酸主要采用有机合成方法进行制备,并采用大型制备液相色谱进行分离提纯,设备价格昂贵且需要消耗大量高纯度有机溶剂,纯化效率差且会产生有毒的有机废液,这也被认为是修饰核苷酸价格居高不下的主要原因。因而,研究三磷酸核苷酸的纯化方法迫在眉睫。
技术实现要素:
5.为了解决现有技术中三磷酸核苷酸化合物纯化方法昂贵,底物适用性不高等问题,本发明提供了一种三磷酸核苷酸盐的制备方法。该方法通过使用阴离子交换柱及三乙胺碳酸盐缓冲液的方法成功实现了多种修饰及非修饰三磷酸核苷酸的大规模自动化制备,无需大量使用有机溶剂和昂贵的制备液相色谱,极大的降低了三磷酸核苷酸的制备成本,并且制备纯度高达99%以上,通用性好,分离度高。
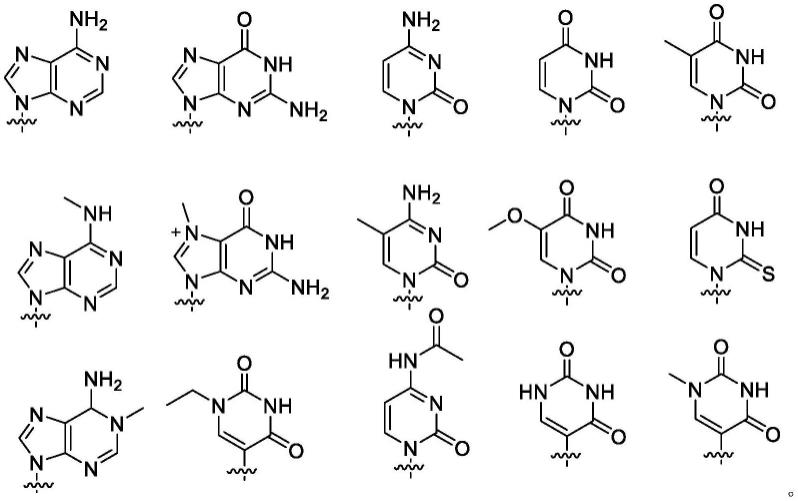
6.本发明提供了一种三磷酸核苷酸三乙胺盐的制备方法,其包括如下步骤:采用色谱法将活性成分为如式c所示的三磷酸核苷酸的待纯化物进行分离制备,得到如式d所示的三磷酸核苷酸三乙胺盐;
300mm,又例如200-220mm。
20.方案一中,所述阴离子交换柱填料的粒径为10-30μm,例如15μm。
21.方案二中,所述阴离子交换柱填料的粒径为40-120μm,例如50-90μm。
22.所述阴离子交换柱的柱温可为4-20℃,例如4℃。
23.所述流动相的起始浓度小于将三磷酸核苷酸三乙胺盐洗脱时的浓度。
24.方案一中,所述buffer a为超纯水。
25.方案一中,所述buffer b为1-2m的三乙胺-碳酸缓冲溶液。
26.方案一中,所述分离制备包括如下步骤:(1)3-5倍柱体积的a buffer平衡;(2)1-3倍柱体积a buffer洗杂,例如2-2.5倍柱体积;(3)5-20倍柱体积洗脱,b buffer的最终比例为18%-80%;例如5.5-8倍柱体积洗脱,b buffer的最终比例为18%-30%;又例如6.5倍柱体积洗脱,b buffer的最终比例为20-30%;(4)1-2倍柱体积洗脱剩余杂质,b buffer比例为50%-100%。
27.方案二中,所述分离制备包括如下步骤:(1)3-5倍柱体积的a buffer平衡;(2)0.5-2倍柱体积a buffer洗杂;(3)8~20倍柱体积洗脱,b buffer的最终比例为12%~100%;(4)1~2倍柱体积洗脱剩余杂质,b buffer的比例为60%~100%。
28.所述洗脱可包括线性梯度洗脱和/或阶梯梯度洗脱,例如以线性梯度的方式逐渐提高b buffer比例,收集色谱峰对应样品溶液。
29.方案一中,当洗脱采用线性梯度洗脱和阶梯梯度洗脱结合时,其可包括以下步骤:以阶梯梯度洗脱的方式初步洗脱杂质,其中b buffer的比例为20%,再以线性梯度的方式逐渐提高b buffer比例,b buffer的比例为30%。
30.方案二中,当洗脱采用线性梯度洗脱和阶梯梯度洗脱结合时,其包括以下步骤:以阶梯梯度洗脱的方式初步洗脱杂质,其中b buffer的比例为12%,再以线性梯度的方式逐渐提高b buffer比例,其中b buffer的比例为30%。
31.所述分离制备可采用下述方法中的任意一种:
32.方法101:以nanoq为填料进行分离制备,以超纯水为buffer a,以1m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)20倍柱体积线性梯度洗脱,b buffer的最终比例为80%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为100%;
33.方法102:以uniq为填料进行分离制备,以超纯水为buffer a,以1m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)20倍柱体积线性梯度洗脱,b buffer的最终比例为80%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为100%;
34.方法103:以generik mc-q为填料进行分离制备,以超纯水为buffer a,以1m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)20倍柱体积线性梯度洗脱,b buffer的最终比例为80%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为100%;
35.方法104:以generik mc-q为填料进行分离制备,以超纯水为buffer a,以2m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)8倍柱体积线性梯度洗脱,b buffer的最终比例为
22.5%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为50%;
36.方法105:以generik mc-q为填料进行分离制备,以超纯水为buffer a,以2m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)5.5倍柱体积线性梯度洗脱,b buffer的最终比例为22.5%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为50%;
37.方法106:以generik mc-q为填料进行分离制备,以超纯水为buffer a,以2m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2.5倍柱体积a buffer洗杂;(3)0.5倍柱体积阶梯梯度洗脱,b buffer的比例为20%,再用6倍柱体积线性梯度洗脱,b buffer的最终比例为30%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为100%;
38.方法107:以generik mc-q为填料进行分离制备,以超纯水为buffer a,以2m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)8倍柱体积线性梯度洗脱,b buffer的最终比例为22.5%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为50%;
39.方法108:以generik mc-q为填料进行分离制备,以超纯水为buffer a,以2m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)8倍柱体积线性梯度洗脱,b buffer的最终比例为18%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为50%;
40.方法201:以deae-sephadex a250为填料,以10mm的三乙胺-碳酸缓冲溶液为buffer a,以1m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)1倍柱体积阶梯梯度洗脱,b buffer比例为12%;再以8倍柱体积线性梯度洗脱,b buffer的最终比例为30%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为60%或100%;
41.方法202:以capto deae为填料,以10mm的三乙胺-碳酸缓冲溶液为buffer a,以1m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)20倍柱体积线性梯度洗脱,b buffer的最初比例为10%,最终比例为100%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为100%;
42.方法203:以unigel-deae为填料,以10mm的三乙胺-碳酸缓冲溶液为buffer a,以1m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)20倍柱体积线性梯度洗脱,b buffer的最终比例为100%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为100%;
43.方法204:以deae sepharose 6ff为填料,以10mm的三乙胺-碳酸缓冲溶液为buffer a,以1m的三乙胺-碳酸缓冲溶液为buffer b,所述分离制备包括下述步骤,(1)5倍柱体积的a buffer平衡;(2)2倍柱体积a buffer洗杂;(3)10倍柱体积线性梯度洗脱,b buffer的最初比例为10%,最终比例为100%;(4)1倍柱体积洗脱剩余杂质,b buffer比例为100%。
44.所述的待纯化物在洗脱前可按照本领域常规进行预处理,以符合进样标准。所述的预处理可包括下述步骤:将所述的待纯化物溶于teab溶液获得上样样品。所述的待纯化物在teab溶液中的浓度可为0-100mg/ml,例如10-20mg/ml。
45.所述的待纯化物通过yoshikawa method或ludwig-eckstein method等有机化学合成方法合成。
46.所述的待纯化物的合成可包括下述步骤:
47.(i)在三磷酸甲酯中,将所述如式a所示的化合物与三氯氧磷进行如下所示的磷酰化反应,得到如式b所示的化合物;
48.(ii)将三正丁胺焦磷酸盐((bu3n)2h4p2o7)、三正丁胺(bu3n)与如式b所示的化合物进行如下所示的反应,并加入水进行淬灭反应,得到如式c所示的化合物;
[0049][0050]
其中,r、r’的定义如前所述。
[0051]
所述步骤(i)中,所述的三氯氧磷与所述的如式a所示的化合物的摩尔比可为1-2:1,例如1.5:1。
[0052]
所述步骤(i)中,所述的三磷酸甲酯与所述的如式a所示的化合物的体积质量比可为10-20ml/g,例如14.9ml/g。
[0053]
所述步骤(i)中,所述反应的反应温度可为-10℃-20℃,例如15℃。
[0054]
所述步骤(i)中,所述反应的反应时间可为20min-40min,例如30min。
[0055]
所述步骤(ii)中,所述的水优选为超纯水。
[0056]
所述步骤(ii)中,所述的三正丁胺焦磷酸盐((bu3n)2h4p2o7)与所述的如式b所示的化合物的摩尔比可为(5-10):1,例如5:1。
[0057]
所述步骤(ii)中,所述的三正丁胺与所述的如式b所示的化合物的摩尔比可为(4-8):1,例如6:1。
[0058]
所述步骤(ii)中,所述反应的反应温度可为5℃-20℃,例如15℃。
[0059]
所述步骤(ii)中,所述反应的反应时间可为5min-20min,例如15min。
[0060]
所述三磷酸核苷酸三乙胺盐的制备方法还可包括如下步骤:在洗脱后,收集纯化后的样品溶液,浓缩,冻干。所述的浓缩优选减压浓缩。所述浓缩温度可为30-35℃。所述减压浓缩的压力可为10mbar。
[0061]
本发明还提供了一种三磷酸核苷酸碱金属盐的制备方法,其包括如下步骤:在溶剂中,在高氯酸盐存在下,将上述制备得到的如式d所示的三磷酸核苷酸三乙胺盐通过如下所示的离子交换反应,制备得到如e所示的三磷酸核苷酸碱金属盐;
[0062]
[0063]
其中m
+
为li
+
或na
+
,取代基r、r’的定义如前所述。
[0064]
所述离子交换反应中,所述的溶剂可为丙酮或乙醇,例如丙酮。
[0065]
所述离子交换反应中,所述的高氯酸盐可为高氯酸锂或高氯酸钠。
[0066]
所述离子交换反应中,所述的高氯酸盐与所述的如式d所示的化合物的摩尔比可为(1000-4000):1,例如1500:1。
[0067]
所述离子交换反应中,所述的溶剂与所述的如式d所示的化合物的体积质量比可为30-100ml/g,例如40ml/g。
[0068]
所述离子交换反应的反应温度可为0℃-25℃,例如4℃。
[0069]
所述离子交换反应的反应时间可为20min-60min,例如30min。
[0070]
所述离子交换反应还可包括如下后处理步骤:过滤,滤饼用溶剂冲洗,干燥。所述的溶剂可为丙酮,优选为4℃的丙酮。所述干燥优选减压干燥。
[0071]
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0072]
本发明所用试剂和原料均市售可得。
[0073]
本发明的积极进步效果在于:(1)实现了多种修饰及非修饰三磷酸核苷酸的大规模自动化制备,通用性好,分离度高,针对不同种类的修饰核苷酸都可以使用此方法进行大规模生产。
[0074]
(2)无需大量使用有机溶剂和昂贵的制备液相色谱,极大的降低了三磷酸核苷酸的制备成本。
附图说明
[0075]
图1为化合物c13以deae-sephadex a250为填料进行分离制备的图谱。
[0076]
图2为化合物c14以capto deae为填料进行分离制备的图谱。
[0077]
图3为化合物c14以unigel-deae为填料进行分离制备的图谱。
[0078]
图4为化合物c14以deae sepharose 6ff为填料进行分离制备的图谱。
[0079]
图5为化合物c14以nanoq为填料进行分离制备的图谱。
[0080]
图6为化合物c14以uniq为填料进行分离制备的图谱。
[0081]
图7为化合物c14以generik mc-q为填料进行分离制备的图谱。
[0082]
图8为化合物c1以generik mc-q为填料进行分离制备的图谱。
[0083]
图9为化合物c2以generik mc-q为填料进行分离制备的图谱。
[0084]
图10为化合物c3以generik mc-q为填料进行分离制备的图谱。
[0085]
图11为化合物c4以generik mc-q为填料进行分离制备的图谱。
[0086]
图12为化合物c6以generik mc-q为填料进行分离制备的图谱。
[0087]
图13为化合物c7以generik mc-q为填料进行分离制备的图谱。
[0088]
图14为化合物c8以generik mc-q为填料进行分离制备的图谱。
[0089]
图15为化合物c9以generik mc-q为填料进行分离制备的图谱。
[0090]
图16为化合物c10以generik mc-q为填料进行分离制备的图谱。
[0091]
图17为化合物c15以generik mc-q为填料进行分离制备的图谱。
具体实施方式
[0092]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0093]
三磷酸甲酯,三氯氧磷,三正丁胺,三正丁胺焦磷酸盐,二氯甲烷,乙腈,三乙胺,氢氧化钠,乙醇均采购于国药试剂官网。核苷原料a1,a2,a3,a4,a6,a7,a9均采购于毕得医药。a5,a8,a10皆为自合成原料。
[0094]
分离制备用柱填料选用deae-sephadex a250,capto deae系列,unideae系列,deae sepharose6ff,uniq系列,nanoq系列,unicore-deae系列,generik mc-deae,generik mc-q,generik mc-deae等阴离子交换树脂填料。纯化设备采用cytiva akta avant 150,akta pure 150或其它蛋白色谱纯化设备。
[0095]
实施例1三磷酸核苷酸化合物c的制备
[0096][0097]
(当r为基团1时,化合物a称为a1、化合物b称为b1、化合物c称为c1;以此类推,当r为基团n时,化合物a称为an、化合物b称为bn、化合物c称为cn)
[0098]
将不同的核苷化合物a分别溶解于三磷酸甲酯并冷却至10℃,缓慢滴加1.5eq的三氯氧磷后将反应体系升温至15℃,并继续搅拌至反应原料完全消失并转化为中间产物b。随后加入5eq预先冷却至-10℃的三正丁胺焦磷酸盐和6eq三正丁胺,反应体系升温至15℃搅拌至原料b完全转化为目标产物c。用预先冷却至4℃的超纯水萃灭反应后调节ph=6用二氯甲烷萃取三次,将萃取完成后的水相体系进行减压蒸馏浓缩可得产物c的粗产品为淡黄绿色油状物,保存于-80℃。
[0099]
实施例2配置2m teab缓冲液
[0100]
在通风橱内将大量样品瓶置于冰上,加入1.1l hplc级三乙胺,加入4l超纯水,通入二氧化碳气体鼓泡,每10分钟测量一次ph值直至ph=8.5,关闭二氧化碳气体。用2μm特氟龙滤器对2m teab缓冲液进行过滤脱气后,封紧瓶口放置于4℃冰箱保存,使用前将2m teab
原液稀释成为1m,50mm,10mm等不同浓度备用。
[0101]
0.5m氢氧化钠水溶液,20%乙醇溶液用于层析柱清洗与保存。
[0102]
实施例3纯化样品准备
[0103]
将产物c的粗产品配置成10-20mg/ml的10mm teab(ph=8.5)水溶液放置于4℃冰箱保存。
[0104]
实施例4以deae-sephadex a250为填料进行制备
[0105]
色谱柱直径25mm,柱高220mm,deae-sephadex a250填料以交联葡聚糖为基质,配基为二乙胺乙基(deae),粒径为40-120μm,a buffer为10mm teab(ph=8.5),b buffer为1m teab(ph=8.5)。
[0106]
制备过程如下:
[0107]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c13为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以1倍柱体积流动相初步洗脱杂质,b buffer比例为12%;然后以线性梯度的方式在8倍柱体积将b buffer比例提高至30%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例为60%(或100%亦可);最后以1倍柱体积流动相冲洗并平衡柱子,不添加b buffer。由计算可得此填料载量为0.48mg/ml,分离度r=1.46(图1)。
[0108]
实施例5以capto deae为填料进行制备
[0109]
色谱柱直径为10mm,柱高为200mm。capto deae填料以具有葡聚糖表面延伸剂的高度交联琼脂糖为基质,配基为二乙胺乙基(deae),粒径为90μm。a buffer为10mm teab(ph=8.5),b buffer为1m teab(ph=8.5)。制备过程如下:
[0110]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c14为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以20倍柱体积将b buffer比例从10%提高至100%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例为100%。由计算可得此填料载量为0.50mg/ml,分离度r=1.60(图2)。
[0111]
实施例6以unigel-deae为填料进行制备
[0112]
色谱柱直径为10mm,柱高为200mm;unigel-deae填料以聚丙烯酸酯(pmma)为基质,配基为二乙胺乙基(deae),粒径为50μm。a buffer为10mm teab(ph=8.5),b buffer为1m teab(ph=8.5)。制备过程如下:
[0113]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c14为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以10倍柱体积将b buffer比例提高至100%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;收集色谱峰对应样品溶液;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例为100%。由计算可得此填料载量为0.62mg/ml,分离度r=1.33(图3)。
[0114]
实施例7以deae sepharose 6ff为填料进行制备
[0115]
色谱柱直径为10mm,柱高为200mm;deae sepharose 6ff填料以琼脂糖凝胶为基
质,配基为二乙胺乙基(deae),粒径为90μm。a buffer为10mm teab(ph=8.5),b buffer为1m teab(ph=8.5)。制备过程如下:
[0116]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c14为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以10倍柱体积将b buffer比例从10%提高至100%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例为100%。由计算可得此填料载量为0.47mg/ml,分离度r=1.80(图4)。
[0117]
实施例8以nanoq为填料进行制备
[0118]
色谱柱直径为7.7mm,柱高为100mm;nanoq填料以单分散聚苯乙烯-二乙烯基苯(ps/dvb)为基质,配基为季铵基团(quaternary ammonium),粒径为10μm。a buffer为超纯水,b buffer为1m teab(ph=8.5)。制备过程如下:
[0119]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c14为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以20倍柱体积将b buffer比例提高至80%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;收集色谱峰对应样品溶液;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例为100%。由计算可得此填料载量为1.01mg/ml,分离度r=2.26(图5)。
[0120]
实施例9以uniq为填料进行制备
[0121]
色谱柱直径为7.7mm,柱高为100mm;uniq填料以单分散聚苯乙烯为基质,配基为季铵基团(quaternary ammonium),粒径为15μm。纯化过程中a buffer为超纯水,b buffer为1m teab(ph=8.5)。制备过程如下:
[0122]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c14为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以20倍柱体积将b buffer比例提高至80%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;收集色谱峰对应样品溶液;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例为100%。由计算可得此填料载量为0.91mg/ml,分离度r=2.4(图6)。
[0123]
实施例10以generik mc-q为填料进行制备
[0124]
色谱柱直径为10mm,柱高为200mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium),粒径为30μm。a buffer为超纯水,b buffer为1m teab(ph=8.5)。
[0125]
制备过程如下:
[0126]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c14为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以20倍柱体积将b buffer比例提高至80%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例为100%。由计算可得此填料载量为0.87mg/ml,分离度r=2.89(图7)。
[0127]
实施例11以generik mc-q为填料进行制备
[0128]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0129]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c1为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以8倍柱体积将b buffer比例提高至22.5%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=1.90(图8)。
[0130]
实施例12以generik mc-q为填料进行制备
[0131]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0132]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c2为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以5.5倍柱体积将b buffer比例提高至22.5%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以2倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=2.41(图9)。
[0133]
实施例13以generik mc-q为填料进行制备
[0134]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0135]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c3为例),再用2.5倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以0.5倍柱体积的流动相将初步杂质洗脱,b buffer比例20%,然后以6倍柱体积将b buffer比例提高至30%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例100%。由计算可得分离度r=1.98(图10)。
[0136]
实施例14以generik mc-q为填料进行制备
[0137]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0138]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c4为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以8倍柱体积将b buffer比例提高至22.5%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=4.38(图11)。
[0139]
实施例15以generik mc-q为填料进行制备
[0140]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,
配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0141]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c6为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以8倍柱体积将b buffer比例提高至22.5%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=3.66(图12)。
[0142]
实施例16以generik mc-q为填料进行制备
[0143]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0144]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c7为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以8倍柱体积将b buffer比例提高至18%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=2.33(图13)。
[0145]
实施例17以generik mc-q为填料进行制备
[0146]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0147]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c8为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以8倍柱体积将b buffer比例提高至22.5%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=4.05(图14)。
[0148]
实施例18以generik mc-q为填料进行制备
[0149]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0150]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c9为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以8倍柱体积将b buffer比例提高至22.5%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=4.10(图15)。
[0151]
实施例19以generik mc-q为填料进行制备
[0152]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0153]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c
(以化合物c10为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以8倍柱体积将b buffer比例提高至22.5%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=2.4(图16)。
[0154]
实施例20以generik mc-q为填料进行制备
[0155]
色谱柱直径为100mm,柱高为220mm;generik mc-q填料以聚甲基丙烯酸酯为基质,配基为季铵基团(quaternary ammonium)。a buffer为超纯水,b buffer为2m teab(ph=8.5)。制备过程如下:
[0156]
层析柱使用前充分消毒、清洗干净,用5倍柱体积a buffer平衡;然后进样产物c(以化合物c15为例),再用两倍柱体积a buffer洗杂,不添加b buffer。然后进行洗脱阶段:首先以8倍柱体积将b buffer比例提高至22.5%,合成中产生的主要杂质:核苷酸单磷酸盐nmp,核苷酸二磷酸盐ndp会依次被洗脱下来,最终洗脱主峰为目标产物ntp;再以1倍柱体积的流动相将剩余杂质洗脱,b buffer比例50%。由计算可得分离度r=1.67(图17)。
[0157]
实施例21三磷酸核苷酸钠盐的制备
[0158][0159]
收集纯化后的样品溶液后进行旋转蒸发减压浓缩至100-200ml,30-35℃,10mbar。将产物浓缩液进行冻干后,可得到不同三磷酸核苷酸的三乙胺盐。
[0160]
将冻干取得的三磷酸核苷酸的三乙胺盐按1:100(m/v)加入5%(m/v)的高氯酸钠/高氯酸锂丙酮溶液,4℃剧烈搅拌1小时至溶液转化为均匀的白色悬浊液。过滤后用200倍体积的4℃丙酮溶液冲洗滤饼后进行减压干燥。可得三磷酸核苷酸钠/锂盐。
[0161]
化合物e质量鉴定
[0162]
质量鉴定采用仪器为shimadzu i-serious uplc,shimadzu lcms 2040,shim-pack gist 5um c18 4.6x250mm,bruker 400mhz。
[0163]
化合物e1:hplc r
t
=2.030min,99.39%.1h nmr(400mhz,d2o):δ8.57(s,1h),8.40(s,1h),6.09(d,j=5.2hz,1h),4.66(t,j=5.2hz,1h),4.53(t,j=4.5hz,1h),4.48
–
4.24(m,3h).
31
p nmr(400mhz,d2o):δ-15.34(d,j=18.6hz),-15.89(d,j=19.5hz),-27.49(t,j=18.9hz).esi-ms m/z calcd for c
10h15
n5o
13
p3[m-h]-506.0;found.506.0
[0164]
化合物e2:hplc r
t
=2.046min,100%.1h nmr(400mhz,d2o):δ8.24(s,1h),5.92(d,j=5.3hz,1h),4.71(t,j=5.2hz,1h),4.53(h,j=4.6hz,1h),4.36(p,j=3.1hz,1h),4.33
–
4.18(m,2h).
31
p nmr(400mhz,d2o):δ-10.63(d,j=19.0hz),-11.15(d,j=19.6hz),-22.73(t,j=19.1hz).esi-ms m/z calcd for c
10h15
n5o
13
p3[m-h]-522.0;found.522.0.
[0165]
化合物e3:hplc r
t
=2.052min,98.12%.1h nmr(400mhz,d2o):δ8.17(d,j=7.9hz,2h),6.30(d,j=7.9hz,2h),5.94(d,j=3.7hz,2h),4.41
–
4.19(m,9h),3.64(q,j=7.1hz,1h),1.17(t,j=7.1hz,2h).
31
p nmr(400mhz,d2o):δ-10.70(d,j=18.9hz),-11.32(d,j=19.5hz),-22.88(t,j=19.1hz).esi-ms m/z calcd for c9h
15
n3o
14
p3[m-h]-482.0;found.482.0.
[0166]
化合物e4:hplc r
t
=2.012min,100%.1h nmr(400mhz,d2o):δ7.97(d,j=8.1hz,1h),6.00(t,j=6.7hz,2h),4.41(t,j=4.5hz,2h),4.30(t,j=3.0hz,1h),4.29
–
4.19(m,2h),3.66(q,j=7.1hz,1h),1.19(t,j=7.1hz,1h).
31
p nmr(400mhz,d2o):δ0.11,-10.55(d,j=19.0hz),-11.31(d,j=19.9hz),-22.87(t,j=19.4hz).esi-ms m/z calcd for c9h
15
n2o
15
p3[m-h]-483.0;found.482.9.
[0167]
化合物e6:hplc r
t
=2.112min,100%.1h nmr(400mhz,d2o):δ2.92(s,3h),4.05(m,2h),4.21(m,1h),4.41(m,1h),4.60(m,1h),5.95(d,1h,j=6.0hz),8.09(s,1h),8.29(s,1h).
31
p nmr(400mhz,d2o):δ
–
21.47(t),
–
10.22(d),7.47(d).esi-ms m/z calcd for c
11h18
n5o
13
p3[m-h]-520.0;found.519.9.
[0168]
化合物e7:hplc r
t
=2.173min,99.38%.1h nmr(400mhz,d2o):δ9.26(s,1h),6.11(d,j=3.6hz,1h),4.73(d,j=4.3hz,1h),4.59(t,j=5.1hz,1h),4.42(d,j=23.2hz,2h),4.31(dd,j=12.6,5.2hz,1h),4.17(s,3h).
31
p nmr(400mhz,d2o):δ-10.44(d,j=19.8hz),-11.39(d,j=20.1hz),-22.91(t,j=19.7hz).esi-ms m/z calcd for c
11h17
n2o
15
p3[m-h]-536.0;found.535.8.
[0169]
化合物e8:hplc r
t
=2.166min,99.38%.1h nmr(400mhz,d2o):δ7.81(s,1h),6.02(d,j=4.7hz,1h),4.46(t,j=4.9hz,1h),4.40
–
4.21(m,4h),2.02(s,3h).
31
p nmr(400mhz,d2o):δ-6.39(d,j=19.8hz),-11.28(d,j=19.0hz),-21.77(t,j=19.8hz).esi-ms m/z calcd for c
11h17
n2o
15
p3[m-h]-496.0;found.495.9.
[0170]
化合物e9:hplc r
t
=2.142min,99.73%.1h nmr(400mhz,d2o):δ7.42(s,1h),6.06(d,j=6.0hz,1h),4.54
–
4.46(m,2h),4.35
–
4.28(m,2h),4.21(dt,j=12.3,3.2hz,1h),3.83(s,3h).
31
p nmr(400mhz,d2o):δ-6.09(d,j=19.4hz),-11.35(d,j=19.8hz),-21.70(t,j=19.7hz).esi-ms m/z calcd for c
11h17
n2o
15
p3[m-h]-514.0;found.513.9.
[0171]
化合物e10:hplc r
t
=2.122min,100%.1h nmr(400mhz,meod):δ4.26(m,2h),4.28(m,1h),4.34(m,1h),4.41(m,1h),6.25(d,1h,j=8.19hz),6.68(d,1h,j=2.83hz),8.23
t
ndp
)/(w
ntp
+w
ndp
)计算产物三磷酸核苷酸ntp与相邻主要杂质ndp的总分离效能指标。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1