一种同时实现囊胚非整倍体检测和高着床潜能筛选的方法
1.本发明涉及医学检测领域,更为具体的,本发明涉及一种囊胚非整倍体检测和高着床潜能筛选的方法。
背景技术:
2.体外受精及胚胎移植是解决临床不孕不育的一个重要手段,通过在体外得到胚胎,并挑选优质胚胎移植从而为不孕不育患者得到后代。在此过程中,临床中结合胚胎着床前遗传学检测(preimplantation genetic testing,pgt)技术和胚胎形态学检查,筛选染色体整倍体及发育形态较好的胚胎进行移植,可以用于提高临床妊娠率,得到更好的临床结局。
3.根据检测类型的不同,目前pgt主要分为胚胎着床前非整倍体检测(preimplantation genetic testing for aneuploidy,pgt-a,也叫拷贝数变异(copy number variation,cnv)检测)、胚胎着床前单基因病检测(preimplantation genetic testing for monogenic diseases,pgt-m)和胚胎着床前染色体结构变异检测(preimplantation genetic testing for chromosomal structural rearrangements,pgt-sr)。
4.pgt-a是临床中最常用的一种染色体筛查技术,可以对胚胎染色体进行整倍性检测,选择染色体数目正常的胚胎进行移植,从而提高临床妊娠率和活产率,该技术此前被称为“pgs
”ꢀ
(preimplantation genetic screening)。在临床体外受精的患者中,高龄(年龄》35岁)及反复移植失败患者胚胎的非整倍体比例较高,对于这些患者pgt-a具有重要意义。目前临床中pgt-a技术主要通过基因组测序或基因组杂交芯片检测胚胎染色体数目,利用甲基化等表观遗传层面检测染色体数量的方法相对匮乏。随着单细胞测序技术的发展与进步,已有研究对人早期胚胎发育各时期的表观遗传组进行了探索,发现早期胚胎发育中发生广泛的表观遗传重编程和转录调控,可控制胚胎发育和后续着床,但是这些研究缺乏胚胎移植及后续发育相关信息,无法使用这些数据对胚胎后续着床及发育结局进行预测。
5.在进行pgt-a得到整倍体胚胎后,一般需要进一步根据胚胎形态学挑选形态较好的胚胎移植。胚胎形态学检查是目前评估胚胎着床及发育潜能的一个常用指标。然而,即使经过pgt-a和形态学筛选的胚胎,在移植后也存在30%
ꢀ‑ꢀ
50%的妊娠失败率,无法高效地挑选出可在母体子宫着床及发育的优质胚胎。因此对胚胎着床及后续发育潜能的预测具有重要临床意义。
6.专利cn105543339b公布了一种同时完成基因位点、染色体及连锁分析的诊断方法。该专利对突变位点周围的snp位点根据人群杂合度进行挑选,使用多重pcr预扩增挑选的snp位点,然后将多重pcr预扩增产物和胚胎扩增产物混合后构建二代测序文库。进行二代测序后,使用全基因组测序的数据进行pgt-a,使用突变位点周围的snp位点进行连锁分析。该技术可以很好地对胚胎的整倍性及变异位点进行检测,但是缺乏对胚胎发育潜能的预测,无法挑选高着床和发育潜能胚胎。文章https://genome.cshlp.org/content/early/
2019/09/23/gr.252981.119中构建了一种可以同时评估胚胎染色体核型和发育潜能的方法。该文章结合囊胚转录组信息与胚胎染色体核型、胚胎形态和胚胎动力学,以整倍体胚胎和形态优良胚胎为高发育潜能胚胎,构建预测模型。对囊胚滋养层细胞进行转录组测序(rna-seq),获得基因表达信息,推导胚胎染色体核型和其发育潜能。该方法通过转录组推断染色体整倍性,由于囊胚细胞中转录组变异较大且不均匀,该方法在用于实现pgt-a染色体非整倍体检测中准确率较低。此外,该预测方法的构建中仅根据核型、胚胎形态及动力学,缺乏临床中真正胚胎移植信息,没有胚胎移植结局,没有在实践中进行应用,实际预测效果未知,具有一定的局限性。专利cn105861658a(所对应文章为https://linkinghub.elsevier.com/retrieve/pii/s1673-8527(17)30140-6)公布了一种通过囊胚甲基化的整体水平高低来区别优质胚胎和非优质胚胎的方法。该方法通过囊胚活检获得滋养层细胞,并对活检的滋养层细胞进行甲基化测序,得到胚胎的整体甲基化水平。并结合加纳德形态囊胚分级镜下筛选形态优良囊胚(形态学评级aa),以形态学评级为aa的优良囊胚的甲基化水平为标准(其认为形态学aa评级的囊胚发育潜能更高),建立高发育潜能胚胎的甲基化数据模型并同时根据甲基化水平筛选高发育潜能的胚胎进行移植。该发明只是以形态学评级为aa的胚胎作为标准参考,认为其甲基化水平预示高的着床和发育潜能,但是临床上很多ab、ba、bb评级的胚胎,仍然可以顺利着床和发育。这种方法局限了胚胎的应用,浪费了很多实际具有着床和发育潜能的胚胎。
7.因此目前在临床中,仍然缺乏一种可以同时进行pgt-a染色体整倍性检测和高着床及发育潜能胚胎筛选的方法。
技术实现要素:
8.为了填补现有技术的空白,本发明提供一种同时对染色体整倍体和优良发育潜能囊胚筛选的检测方法,该方法使用单细胞甲基化测序实现囊胚着床前染色体非整倍性遗传学检测和囊胚发育潜能预测,挑选染色体整倍体及高着床潜能囊胚用于后续移植。为了实现上述技术效果,本发明具体提供如下的技术方案:本发明的第一个方面,提供一种同时实现囊胚非整倍体检测(pgt-a)和高着床潜能筛选的方法,所述方法包括如下步骤:1. 获取1-3个囊胚滋养层细胞作为检测样本;2. 裂解胚囊滋养层细胞,获得细胞内dna;3. 将dna进行亚硫酸氢盐转化;4. 亚硫酸氢盐转化后代的dna产物文库构建;5. 对构建的甲基化文库进行测序,得到胚胎甲基化数据;6. 数据分析及着床潜能预测:(1)去除甲基化数据测序接头、低质量碱基、过短的序列;(2)使用人类参考基因组作为对照,使用甲基化比对软件将上一步所得序列与人类参考基因组进行比对,比对后所得bam文件,去除非唯一比对序列及重复序列,将处理后的bam文件使用readcounter对基因组每个位置的序列数进行计数,得到每个位置的序列数,然后使用hmmcopy软件进行cnv计算,得到每个样本的cnv结果,实现pgt-a;(3)使用bismark_extractor计算每个样本在每个覆盖到的cg位点的甲基化水平,
进一步将样本全基因组从5’端-3’端顺序划分为连续的序列检测窗口区域,每个窗口区域为300bp序列长度,计算每个样本在挑选的11个窗口区域的甲基化水平;(4)使用11个窗口区域的甲基化水平作为变量x,权重系数记为w,将二者代入着床概率函数:概率函数,其中,x=[x1,x2,
…
,x11]为自变量矩阵;w=[w1,w2,
…
,w11]为权重系数矩阵,权重系数由python软件中的sklearn.linear_model的logisticregression函数对临床获得的多个着床成功以及着床失败的囊胚样本拟合计算获得;z=wx+w0;(5)判断标准:如果概率大于0.5,则判定胚胎可以着床。
[0009]
在一种实施方式中,上述步骤4在文库构建前先将全基因组进行预扩增及扩增产物纯化。
[0010]
在一种实施方式中,上述步骤5所述测序为通过使用测序仪来进行测序。
[0011]
在一种实施方式中,上述步骤6(1)中所用软件为trim_galore。
[0012]
在一种实施方式中,上述步骤6(2)中所用甲基化比对软件为bismark。
[0013]
在一种实施方式中,上述步骤6(3)中所述11个序列检测窗口为:chr14:58255501-58255800、chr16:69220201-69220500、chr17:77496001-77496300、chr19:17769301-17769600、chr19:54994501-54994800、chr1:32130901-32131200、chr22:47130901-47131200、chr2:30944701-30945000、chr2:39253201-39253500chr3:105114001-105114300、chr6:138312301-138312600。
[0014]
在一种实施方式中,上述步骤6(4)中所述权重系数矩阵为w=[-0.7301037860241746,-0.7632086871621979,-0.719178146569863,0.6693682018249012,-0.6606863750100334,0.7006178980344422,-0.6718163228307065,0.7963020595213105,-0.6394799519724385,-0.7478914784023284,-0.6883953758940006]。
[0015]
相对于现有技术本发明获得了如下显著的进步:1.本发明中公开的方法不仅可以检测全基因组的拷贝数变异,完成pgt-a,还可以实现着床潜能的预测,挑选到高着床潜能胚胎进一步移植,增加了该项重要功能;2.本发明提供的方法适用于更加广泛的囊胚,现有检测方法通常仅仅使用aa评级囊胚,损失了众多具有着床潜能的ab、ba、bb胚胎,本发明所述方法囊括了该部分胚胎,很大程度提高了囊胚的利用率;3.本发明首次纳入了临床中真实的囊胚移植后妊娠结局,预测模型与真实情况更加接近,在pgt-a和胚胎发育潜能预测中均具有较高的成功率,在实际临床应用中也取得良好效果。
附图说明
[0016]
附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:
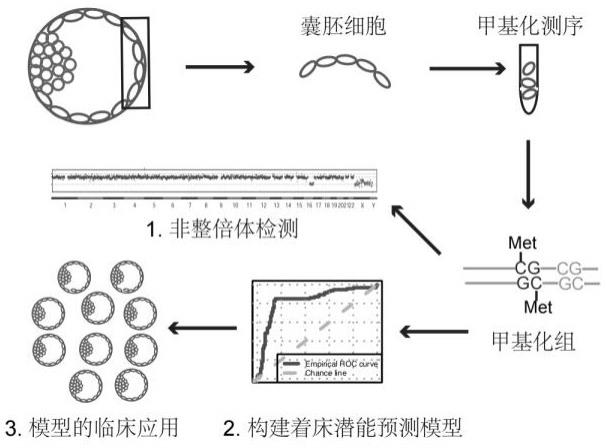
图1为本发明技术路线图;图2为1号囊胚e1囊胚滋养层细胞样本甲基化文库质控结果;图3为2号囊胚e2囊胚滋养层细胞样本甲基化文库质控结果;图4为3号囊胚e3囊胚滋养层细胞样本甲基化文库质控结果;图5为每个样本的pgt-a结果图。
具体实施方式
[0017]
以下结合附图对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
[0018]
实施例1 检测方法的构建本发明检测方法技术路线如图1所示。具体描述如下:1. 囊胚滋养层细胞获得:临床中行辅助生殖技术助孕的夫妇,卵母细胞通过单精子注射方式获得受精卵,近一步培养发育至囊胚。行常规囊胚滋养层活检,对活检下来的滋养层细胞团块,使用混合酶(accutase:胰酶 = 1:1)在37℃反应50-60min,消化为单细胞。选取1-3个单细胞作为检测样本。
[0019]
2. 细胞裂解:用口吸管将1-3个消化获得的滋养层细胞吹入加入5 μl甲基化裂解液(0.1 μl 1 m tris-edta,0.1 μl 1 m kcl,0.15 μl 10% triton-x 100,0.25 μl 20 mg/ml 蛋白酶,1 μl 10 ng/μl carrier rna,0.6 pg lambda dna,使用nf水补齐)的pcr管中,放入pcr仪中裂解细胞,释放dna;程序:(1)50℃反应3h;(2)75℃反应30min。
[0020]
3. 亚硫酸氢盐转化:使用dna methylation-directtm magprep试剂盒完成基因组dna亚硫酸氢盐转化实验及纯化,使用19.5μl nf水洗脱样本。
[0021]
4. 全基因组预扩增:4.1 一链合成反应体系:洗脱的19.5 μl的基因组dna 中加入2.5 μl 10
×ꢀ
neb buffer 2,1 μl 10 μm oligo1(5
′‑
ctacacgacgctcttccgatctnnnnnn-3
′
)和1 μl dntp(10 mm each)程序:第1轮:(1)65℃反应3 min;(2)每个pcr管中,冰上加入1 μl klenow exo-(50 u/μl,neb);(3)4℃反应5 min,+1
°
c/15 s 至 37℃;(4)37℃反应30 min;第2-6轮:(1)95℃反应1 min
(2)每个pcr管中,冰上加入2 μl 1
×ꢀ
neb buffer 2和0.5 μl klenow exo-(50 u/μl,neb);(3)4℃反应5 min,+1℃/15 s 至 37
°
c;(4)37℃反应30 min;(5)重复(1)到(4)步骤5个循环。
[0022]
4.2 消化多余引物每个pcr管中加入2 μl exonuclease i(20 u/μl,neb),pcr仪中37℃孵育1 h。
[0023]
4.3 产物纯化(1)向dna产物中,加入0.8
×ꢀ
ampure xp beads;(2)室温孵育10min;(3)80%乙醇洗两次;(4)使用39 μl nf水洗脱。
[0024]
4.4 二链合成:反应体系:向每个pcr管中加入5
ꢀµ
l 10
×ꢀ
neb buffer 2,2
ꢀµ
l 10
ꢀµ
m oligo2(5
′‑
agacgtgtgctcttccgatctnnnnnn-3
′
)和2
ꢀµ
l dntp(10 mm each);程序:(1)95℃反应45 s;(2)每个pcr管中,冰上加入2
ꢀµ
l klenow exo-(50 u/
µ
l,neb);(3)4℃反应5 min,+1℃/15 s to 37℃;(4)37℃反应60 min。
[0025]
4.5 产物纯化:(1)向dna产物中,加入0.8
×ꢀ
ampure xp beads;(2)室温孵育10min;(3)80%乙醇洗两次;(4)使用22 μl nf水洗脱。
[0026]
5. 全基因组扩增产物文库构建:洗脱的dna转移至一个新的pcr管中,加入1.5
ꢀµ
l 10
ꢀµ
m universal pcr primer(neb),1.5
ꢀµ
l 10
ꢀµ
m pcr index primer(neb)和25
ꢀµ
l 2
×ꢀ
kapa hifi hotstart readymix;程序:(1)95℃反应3min;(2)98℃反应20s;(3)65℃反应30s;(4)72℃反应1min;(5)72℃反应1min;(6)重复(2)到(4)步骤16个循环。
[0027]
6. 产物纯化两次:(1)向dna产物中,加入0.8
×ꢀ
ampure xp beads;
(2)室温孵育10min;(3)80%乙醇洗两次;(4)使用25 μl nf水洗脱;(5)重复(1)到(4)步骤一次。
[0028]
7. 使用测序仪对构建的甲基化文库进行测序,得到胚胎甲基化数据。
[0029]
8. 数据比对及pgt-a(1)使用trim_galore去除甲基化数据测序接头、低质量碱基、过短的序列。
[0030]
(2)将上一步处理所得数据使用甲基化比对软件bismark比对到人类参考基因组,比对后所得bam文件,去除非唯一比对序列及重复序列,将处理后的bam文件使用readcounter对基因组每个位置的序列数进行计数,得到每个位置的序列数,然后使用hmmcopy软件进行cnv计算,得到每个样本的cnv结果。
[0031]
9. 构建着床潜能预测模型(1)使用bismark_extractor计算每个样本在每个覆盖到的cg位点的甲基化水平。进一步将样本全基因组从5’端-3’端顺序划分为连续的序列检测窗口,每个窗口为300bp序列长度,计算每个样本的每个窗口的甲基化水平。
[0032]
(2)为构建着床潜能预测模型,以临床中获得的21个着床成功和18个着床失败囊胚的滋养层细胞甲基化测序结果为数据集。为寻找可以指示胚胎着床潜能的基因组标记,首先计算着床成功组和着床失败组的差异甲基化区域。具体为:对于基因组每一个300bp区域,使用非配对双尾 t 检验计算着床成功组和着床失败组的甲基化差异值和统计显著性。选取差异值≥0.1,p值小于0.01的区域作为差异甲基化区域。一共得到1198个在着床成功囊胚中显著高甲基化和2441个在着床成功囊胚中显著低甲基化区域。
[0033]
(3)将上一步所得差异甲基化区域,使用python sklearn.feature_selection.rfecv的递归特征减少法挑选最优的可区分着床成功和失败的差异甲基化区域,最终得到11个区域,这11个区域的甲基化水平可以很好地指示囊胚的着床结局(如表1所示)。
[0034]
表1:递归特征减少法筛选所得11个高指示度区域
区域1区域2区域3区域4区域5区域6区域7区域8区域9区域10区域11chr14:58255501-58255800chr16:69220201-69220500chr17:77496001-77496300chr19:17769301-17769600chr19:54994501-54994800chr1:32130901-32131200chr22:47130901-47131200chr2:30944701-30945000chr2:39253201-39253500chr3:105114001-105114300chr6:138312301-138312600
进一步,使用上述21个着床成功和18个着床失败囊胚作为样本,使用表1中所示的11个区域的甲基化水平作为变量,构建logistic回归模型。具体为:设定x=[x1, x2,
ꢀ…
, x11]为自变量矩阵,w=[w1, w2,
ꢀ…
, w11]为权重系数矩阵,则得到线性函数 z=wx+w0。进一步得到概率函数。 其中权重系数矩阵w由此前临床获得的21个着床成功和18个着床失败囊胚样本拟合获得,矩阵为w=[-0.7301037860241746,
ꢀ‑
0.7632086871621979,
ꢀ‑
0.719178146569863, 0.6693682018249012,
ꢀ‑
0.6606863750100334, 0.7006178980344422,
ꢀ‑
0.6718163228307065, 0.7963020595213105,
ꢀ‑
0.6394799519724385,
ꢀ‑
0.7478914784023284,
ꢀ‑
0.6883953758940006]。
[0035]
(4)针对新获得囊胚,使用上一步所得概率模型,计算囊胚着床概率,如果概率大于0.5,则判定胚胎可以着床,临床上优先挑选高着床概率囊胚移植。
[0036]
实施例2 利用所述方法进行囊胚筛选本家系包括一对经历过2次囊胚移植后着床失败的夫妇,其最近辅助生殖周期一共得到3枚囊胚。该夫妇期望选择1个染色体整倍体且着床潜能高的囊胚移植。增加妊娠成功率。
[0037]
1. 囊胚滋养层细胞获得:对这对夫妇临床体外受精中获得的3枚囊胚,分别进行常规囊胚外滋养层活检。得到的滋养层细胞团块,使用混合酶(accutase:胰酶 = 1:1)在37℃反应50-60min,消化为单细胞,分别选取:1号囊胚e1:评级为5ab,活检获得1个细胞+2个小细胞2号囊胚e2:评级为5aa,活检获得2个细胞3号囊胚e3:评级为5bb,活检获得2个细胞作为检测样本。
[0038]
2. 细胞裂解:用口吸管将滋养层细胞吹入加入5 μl甲基化裂解液(0.1 μl 1 m tris-edta,0.1 μl 1 m kcl,0.15 μl 10% triton-x 100,0.25 μl 20 mg/ml 蛋白酶,1 μl 10 ng/μl carrier rna,0.6 pg lambda dna,使用nf水补齐)的pcr管中,放入pcr仪中裂解细胞,释放dna;程序:(1)50℃反应3h;(2)75℃反应30min。
[0039]
3. 亚硫酸氢盐转化:使用dna methylation-directtm magprep试剂盒完成基因组dna亚硫酸氢盐转化实验及纯化,使用19.5μl nf水洗脱样本。
[0040]
4. 全基因组预扩增:4.1 一链合成反应体系:洗脱的19.5 μl的基因组dna 中加入2.5 μl 10
×ꢀ
neb buffer 2,1 μl 10 μm oligo1(5
′‑
ctacacgacgctcttccgatctnnnnnn-3
′
)和1 μl dntp(10 mm each)程序:第1轮:(1)65℃反应3 min;(2)每个pcr管中,冰上加入1 μl klenow exo-(50 u/μl,neb);(3)4℃反应5 min,+1
°
c/15 s 至 37℃;(4)37℃反应30 min;第2-6轮(1)95℃反应1 min;(2)每个pcr管中,冰上加入2 μl 1
×ꢀ
neb buffer 2和0.5 μl klenow exo-(50 u/μl,neb);
(3)4℃反应5 min,+1℃/15 s 至 37
°
c;(4)37℃反应30 min;(5)重复(1)到(4)步骤5个循环。
[0041]
4.2 消化多余引物每个pcr管中加入2 μl exonuclease i(20 u/μl,neb),pcr仪中37℃孵育1 h。
[0042]
4.3 产物纯化(1)向dna产物中,加入0.8
×ꢀ
ampure xp beads;(2)室温孵育10min;(3)80%乙醇洗两次;(4)使用39 μl nf水洗脱。
[0043]
4.4 二链合成反应体系:向每个pcr管中加入5
ꢀµ
l 10
×ꢀ
neb buffer 2,2
ꢀµ
l 10
ꢀµ
m oligo2(5
′‑
agacgtgtgctcttccgatctnnnnnn-3
′
)和2
ꢀµ
l dntp(10 mm each);程序:(1)95℃反应45 s;(2)每个pcr管中,冰上加入2
ꢀµ
l klenow exo-(50 u/
µ
l,neb);(3)4℃反应5 min,+1℃/15 s to 37℃;(4)37℃反应60 min。
[0044]
4.5 产物纯化(1)向dna产物中,加入0.8
×ꢀ
ampure xp beads;(2)室温孵育10min;(3)80%乙醇洗两次;(4)使用22 μl nf水洗脱。
[0045]
5. 全基因组扩增产物文库构建;洗脱的dna转移至一个新的pcr管中,加入1.5
ꢀµ
l 10
ꢀµ
m universal pcr primer(neb),1.5
ꢀµ
l 10
ꢀµ
m pcr index primer(neb)和25
ꢀµ
l 2
×ꢀ
kapa hifi hotstart readymix;程序:(1)95℃反应3min;(2)98℃反应20s;(3)65℃反应30s;(4)72℃反应1min;(5)72℃反应1min;(6)重复(2)到(4)步骤16个循环。
[0046]
6. 产物纯化两次(1)向dna产物中,加入0.8
×ꢀ
ampure xp beads;(2)室温孵育10min;(3)80%乙醇洗两次;
(4)使用25 μl nf水洗脱;(5)重复(1)到(4)步骤一次。
[0047]
7. 对获得的甲基化文库进行qubit测浓度:表2:3枚囊胚甲基化文库浓度编号浓度(ng/μl)index编号e114.1nebindex1e213.4nebindex2e316.1nebindex38. 对获得的甲基化文库进行片段化分析,得到各囊胚甲基化文库(图2-4),文库片段长度集中在350-380bp,没有小片段污染,质控合格,进行二代测序,得到各囊胚甲基化测序数据。
[0048]
9. 数据分析及着床潜能预测9.1 样本测序数据比对及数据质量评估将测序所得甲基化数据,使用trim_galore去除每个样本测序数据的接头序列、低质量碱基、短序列(《36bp),得到清洗后的数据。
[0049]
将清洗后数据使用bismark比对到人类参考基因组(hg38),并统计每个样本的比对后数据质量,如下表3所示:表3:3枚胚胎比对后序列信息胚胎编号覆盖的cg位点数目重亚硫酸盐转化率e111,091,17299.9%e212,749,70299.0%e312,728,60799.8%9.2 pgt-a分析将比对后的bam文件使用readcounter对比对后的基因组每个位置进行计数,得到每个位置的序列数据,然后使用hmmcopy软件进行cnv计算,得到每个样本的cnv结果(图5),实现pgt-a。本实例中,检测到1号囊胚e1在16号染色体长臂具有部分缺失,2号胚胎e2和3号胚胎e3为整倍体囊胚。
[0050]
根据加德纳形态囊胚分级系统,e2囊胚为aa级胚胎,e3囊胚为bb级胚胎,以往临床中会优先移植e2胚胎。
[0051]
9.3 使用甲基化预测模型预测胚胎着床潜能进一步,计算本实施例得到的2枚整倍体胚胎e2、e3在案例1中9(3)所述的11个区域的甲基化水平,如下表4所示:表4:2枚整倍体胚胎在高区分度区域的甲基化水平
胚胎编号区域1区域2区域3区域4区域5区域6区域7区域8区域9区域10区域11e20.742694805000.1224603170.6293290040.2083333330.750.8333333330.56522366510.657592593e30.6000.6113466390.1666666670.684523810.0476190480.66666666700.3333333330.152272727
进一步将这些水平代入前述案例1中9(3)已构建的着床概率预测函数,得到2枚胚胎的着床概率如下表所示:表5:2枚整倍体胚胎着床潜能预测结果胚胎编号着床概率(概率值为0-1)是否可着床
e20.4141804否e30.9064531是实施例3 对比例(根据已有专利cn105861658a分析着床潜能)在本项目中,我们也使用专利cn105861658a所述方法对e2和e3两枚胚胎进行着床潜能分析。
[0052]
采用专利cn105861658a的分析方法对e2和e3两枚胚胎甲基化水平进行分析,显示两枚胚胎的甲基化水平分别为0.30和0.32,均为0 .3
±
0 .2范围内,均符合专利cn105861658a所述的高着床潜能胚胎,无法进一步挑选。本案例中,进一步结合胚胎评级,患者首先选择形态学评级为aa的e2作为移植胚胎。
[0053]
表6:专利cn105861658a所述方法对胚胎着床潜能预测结果胚胎编号甲基化水平胚胎评级专利cn105861658a结果e20.30aa可着床e30.32bb可着床1. 移植后妊娠结果比较:该夫妇首先选择了已有专利cn105861658a的分析方法所得的结果,选择了甲基化水平在0.3
±
0.2范围内且形态学评级为aa的e2胚胎移植,移植14天后血hcg水平12 mui/ml,胚胎未着床。
[0054]
e2胚胎着床失败后,患者选择移植剩余的e3胚胎(本项目预测e3着床潜能高于e2),移植14天后血hcg水平112mui/ml,移植后21天血hcg水平1950mui/ml,移植后40天超声检查宫内可见妊娠囊,可探及胎心搏动。确定临床宫内妊娠。
[0055]
2. 综合比较结果:根据专利cn105861658a提供的分析方法,筛选得到e2及e3胚胎均为染色体整倍体,并且均具有高的着床潜能,进一步,通过形态学评级,aa评级的e2胚胎在临床中应予以优先移植。但实际结果显示,即使甲基化水平在专利cn105861658a所述的0.3
±
0.2范围内且评级aa,也不一定具有更高的着床潜能。
[0056]
根据上述结果可见,本研究主要关注与胚胎着床潜能相关度最高的11个区域,通过这11个区域来预测胚胎着床潜能,结果显示尽管e3胚胎加德纳形态囊胚分级为bb级,但e3胚胎具有更高的着床潜能,在实际移植后也成功妊娠。对该实施例高着床潜能胚胎的成功筛选,证明本研究构建的着床潜能预测模型,在临床应用中真实有效。
[0057]
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1