一类新型可光交联型光敏性材料及其应用
1.本发明属于功能有机高分子材料领域,具体涉及一类新型可光交联型光敏性材料及其应用。
背景技术:
2.聚酰亚胺(pi)作为一种特殊的功能高分子材料,是一类分子主链中含有酰亚胺基团的芳杂环高分子化合物。其聚合物分子链含有芳环和亚胺环,由于酰亚胺环是平面对称的环状结构,其键角和键长都处于稳定的状态,因此聚酰亚胺分子中除了存在经典的范德华力外,还存在电荷转移络合物的优势层间堆砌以及混合层堆砌等,这些结构共同决定了聚酰亚胺不仅刚性大、熔点高、耐热性好、耐化学腐蚀性、机械强度高,还具有耐辐射性,良好的电绝缘性、低介电常数和阻燃性等优异性能。因此,已被广泛应用在半导体、三维打印、光波导材料、液晶材料、非线性光学材料等领域,成为一类应用前景广阔的功能高分子材料
3.聚酰亚胺分子主链上一般含有苯环和酰亚胺环结构,由于电子极化和结晶性致使聚酰亚胺存在较强的分子链间作用,引起聚酰亚胺分子链紧密堆积,从而导致传统的聚酰亚胺既不熔化又不溶解,难以加工。在应用于半导体、三维打印材料领域时,由于缺乏光敏性,使得工艺流程繁琐,制造成本增加,存在一定的局限性。因此,对于既能保留传统聚酰亚胺耐热性、良好电绝缘性等优异性能的同时,又能改善溶解性,增加光敏性的光敏聚酰亚胺(pspi)材料的研究,具有非常重要的意义。
4.螺环芳烃类化合物,因其具有大的共轭体系和独特的螺共轭效应、刚性共平面结构、高的玻璃化转变温度以及良好的热稳定性等优点,成为有机高分子材料的重要结构单元。在螺环芳烃类材料中,螺[芴-9,9
’‑
氧杂蒽]通过中心sp3杂化的c原子连接芴环与氧杂蒽环两部分,并且在不同的位点可以用不同的有机基团进行修饰改性,这样富含电子的氧杂蒽环与芴环的螺环组合结构在分子领域中能有效构建出非平面的三维分子构象。空间位阻效应可以有效的抑制分子间的π-π相互作用,从而改善材料的溶解性与稳定性。因此,研究将该类材料应用于聚酰亚胺去改善溶解性等是很有前景的。
技术实现要素:
[0005]
本发明研究光敏性二胺单体及应用,开发出一类可光交联型光敏性二胺单体,该单体可与不同类型二元酸酐聚合得到不同的可光交联型光敏聚酰亚胺,用作光交联、光固化材料,有望应用但不限于光刻胶、三维打印等领域。
[0006]
本发明首先提供一类新型可光交联型光敏性材料,所述材料具有如下通式z、paa、pspi的结构:
[0007][0008]
其中,x各自独立的选自n为大于1的数;
[0009]
具体的,n为1-1000的数;
[0010]
具体的,n为2-500的数。
[0011]
r1,r2各自独立的选自各自独立的选自m为0-10的整数,r3为含有取代基或者不含取代基的c3-c8的环烯烃。
[0012]
所述取代基选自c1-c20的烷烃、卤素。
[0013]
具体的,r3可选自
[0014]
y各自独立的选自商业化的脂肪族四甲酸二酐、脂环族四甲酸二酐、芳香族四甲酸二酐、杂环四甲酸二酐的母体结构。
[0015]
具体的,y各自独立的选自商业化的c4-c30脂肪族四甲酸二酐、c4-c30脂环族四甲酸二酐、c4-c30芳香族四甲酸二酐、c4-c30杂环四甲酸二酐的母体结构。
[0016]
所述的母体结构是四甲酸二酐上酸酐键以外的结构。
[0017]
具体的,y各自独立的选自具体的,y各自独立的选自具体的,y各自独立的选自
[0018]
具体的,所述一类新型可光交联型光敏性材料可选自如下结构:
[0019][0020]
其中,x,y的定义同结构通式中的定义。
[0021]
通式w所述的可光交联型光敏性二胺单体,以螺[芴-9,9
’‑
氧杂蒽]为核,芴环与氧杂蒽环两个垂直的非共轭单元使其具有三维大体积空间位阻的刚性结构,由其合成的一系列衍生物都具有较高的熔点和玻璃化转变温度,而且能够抑制分子的结晶,具有良好的成膜性,提高分子的热力学稳定性。氧杂蒽环上的氨基、酚羟基通过连接可光交联基团使该单体具备光敏性,芴环的2,7-位具有很好的反应活性,为其结构向三维空间的延伸提供便利。该类可交联型光敏性二胺单体与不同的二元酸酐聚合,得到一系列的光敏聚酰亚胺(如通式paa和pspi所示),可用作光交联、光固化材料,有望应用但不限于光刻胶、三维打印等领域。
[0022]
所述的可光交联型光敏性材料的制备方法,所述方法包括以芴酮和不同取代的苯酚为原料进行螺[芴-9,9
’‑
氧杂蒽]的构建,以氧杂蒽环上的氨基与酚羟基为衍生位点连接可交联型光敏性基团,通过芴环2,7-位双氨基与不同二元酸酐聚合得到不同的光敏聚酰亚胺,具体包括以下步骤:
[0023]
化合物a通过一步硝化反应后得到的化合物b:
[0024][0025]
化合物b与不同取代的苯酚(化合物u)在酸催化下一步成环构建化合物c:
[0026][0027]
化合物c和化合物v通过酰氯与羟基的酯化反应或酰氯与氨基的酰胺化反应得到化合物d:
[0028][0029]
其中,x定义同结构通式中的定义,化合物v选自如下结构:
[0030][0031]
其中,m=0-10的整数,r3为含有3-8个碳的环烯烃及其衍生物;
[0032]
化合物d和zn粉在酸性条件下反应还原硝基,得到化合物w:
[0033][0034]
其中,x的定义同上述结构通式中的定义。
[0035]
化合物w与不同的二元酸酐(化合物k)聚合得到不同的聚酰胺酸paa或者光敏聚酰亚胺pspi:
[0036][0037]
其中,x、y的定义同上述结构通式中的定义。选自商业化的脂肪族四甲酸二酐、脂环族四甲酸二酐、芳香族四甲酸二酐、杂环四甲酸二酐,其中y为所述脂肪族四甲酸二酐、脂环族四甲酸二酐、芳香族四甲酸二酐、杂环四甲酸二酐的母体结构,中的环并不
用于限定y一定为环状结构。
[0038]
具体的,所述的可光交联型光敏性材料的制备方法可以采用以下步骤:
[0039]
1)化合物a加入到少量去离子水中,将混合物加热到80℃,向上述溶液中逐滴滴加浓hno3/浓h2so4的混合液,加热回流1h后,再次逐滴滴加浓hno3/浓h2so4的混合液,继续加热回流反应,混合液逐渐粘稠且呈黄色,反应结束后,冷却,将反应液倒入水中,析出黄色沉淀,抽滤、洗涤、干燥,将干燥后的粗产物在乙醇中重结晶得到纯净的化合物b。
[0040]
2)在惰性气体气氛下,将化合物b与化合物u混合均匀,向混合物中加入对甲苯磺酸,140℃下反应,降温,向反应瓶中加入去离子水,搅拌0.5h,粗产物从反应混合物中沉淀出来,抽滤、干燥为棕色固体,将干燥后的粗产物通过柱层析提纯,得到化合物c;
[0041]
所述化合物b与不同取代的苯酚(化合物u)的投料摩尔比为1:2-10。
[0042]
3)在惰性气体气氛下,将化合物c溶解于干燥的丙酮中,向溶液中滴加三乙胺,搅拌10min后,将化合物v分散到干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,在室温下搅拌,反应结束后向反应液中加入乙酸乙酯,水洗、干燥、柱层析得到化合物d。
[0043]
4)在惰性气体气氛下,将化合物d溶解在水/乙醇(体积比=1/3)的混合体系中,加入还原zn粉、氯化铵和冰乙酸,50℃下搅拌4-6h,抽滤、萃取、干燥、柱层析获得化合物w,
[0044]
所述化合物d与还原zn粉的投料摩尔比例为1:6-15。
[0045]
5)在惰性气体气氛下,将化合物w溶解于干燥且除氧的氮甲基吡咯烷酮中,待完全溶解后,向溶液中加入不同的二元酸酐(化合物k),25℃下反应0.5h以上,将反应液在合适的有机溶剂中沉出、洗涤、干燥,获得不同的光敏聚酰胺酸paa;向聚酰胺酸paa在氮甲基吡咯烷酮的溶液中加入三乙胺、吡啶、甲苯,180℃下脱水环化5h,降温,将反应液在合适的有机溶剂中沉出、洗涤、干燥,获得不同的光敏聚酰亚胺pspi;
[0046]
所述化合物y与不同二元酸酐(化合物k)的投料摩尔比为1:0.95-1.10。
[0047]
步骤1所述的两批浓hno3/浓h2so4的混合液的比例为1:1。
[0048]
步骤2所述的化合物b与不同取代的苯酚(化合物u)的投料摩尔比优选为1:2.5。
[0049]
步骤4所述的化合物d与还原zn粉的投料摩尔比优选为1:12。
[0050]
步骤5所述的化合物w与二元酸酐(化合物m)的投料摩尔比优选为1:1.05。
[0051]
步骤5所述的有机溶剂选自二氯甲烷、乙酸乙酯、乙醇。
[0052]
所述的可光交联型光敏性二胺单体在改善聚酰亚胺的性质方面具有良好的应用效果。
[0053]
所述的可光交联型光敏性二胺单体制备的光敏聚酰亚胺在光交联、光固化材料中具有良好的应用前景。
[0054]
本发明的有益效果:一类新型可光交联型光敏性材料及其应用,这类可光交联型光敏性二胺材料以螺[芴-9,9
’‑
氧杂蒽]为核,芴环与氧杂蒽环两个垂直的非共轭单元使其具有三维大体积空间位阻的刚性结构,氧杂蒽环上的氨基与酚羟基连接可光交联基团使该材料具备光敏性。芴环2,7-位的两个氨基能够与不同的二元酸酐进行聚合,获得具有良好溶解性、成膜性与热力学稳定性的可光交联型光敏聚酰亚胺,该可光交联型光敏聚酰亚胺作为光交联、光固化材料,有望应用但不限于光刻胶、三维打印等领域。
附图说明
[0055]
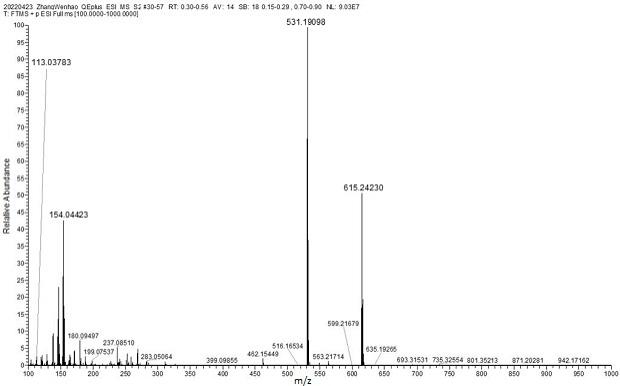
图1为可光交联型二胺单体w-1的高分辨质谱图。
[0056]
图2为可光交联型二胺单体w-1的核磁共振氢谱表征图。
[0057]
图3为可光交联型二胺单体w-1的核磁共振碳谱表征图。
[0058]
图4为paa-1与pspi-1的傅里叶红外光谱表征图。
[0059]
图5为pspi-1紫外光照射前后的傅里叶红外光谱表征图。
具体实施方式
[0060]
下述非限制性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思进行等同替换或改变均属于本发明保护范畴。
[0061]
下述实施例中,如无特殊说明,所用试剂均可由常规方法制备或由商业途径购买。
[0062]
实施例1
[0063]
可光交联型二胺单体(w-1)的合成路线:
[0064][0065]
(1)中间体b的合成
[0066]
氮气气氛中,将a(2.00g,11.10mmol)分散在5ml的去离子水中,将混合物加热到80℃,逐滴滴加浓hno3(65%,6.92ml,99.89mmol)/浓h2so4(98%,6.64ml,112.08mmol)的混合液,加热回流1h后,再次逐滴滴加浓hno3(65%,6.92ml,99.89mmol)/浓h2so4(98%,6.64ml,112.08mmol)的混合液,继续加热回流反应。tlc监测反应结束后,将反应液冷却,倒入冰水中,产生黄色沉淀,抽滤,滤饼洗涤3次后真空干燥,将干燥得到的粗产物在乙醇中重结晶,抽滤获得化合物b。
[0067]
(2)中间体c1的合成
[0068]
氮气气氛中,化合物b(2.00g,7.40mmol)和间苯二酚(2.04g,18.50mmol)混合均匀,将对甲苯磺酸(2.55g,14.80mmol)加入到混合物中,140℃下干粉反应,tlc监测反应结束后,降温,向反应瓶中加入15ml去离子水,搅拌0.5h,粗产物从反应混合物中沉淀出来,抽滤、洗涤、干燥后为棕色固体,将干燥后的粗产物通过柱层析(甲醇/二氯甲烷体系=1/50)分离提纯获得黄色固体化合物c1;
[0069]
(3)中间体d1的合成
[0070]
氮气气氛下,化合物c1(1.00g,2.20mmol)溶解于8ml干燥的丙酮中,向溶液中滴加三乙胺(672.99mml,4.84mmol),搅拌10min后,将甲基丙烯酰氯(468.62mml,4.84mmol)分散到2ml干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,加入催化剂量的dmap催化反应
进行,在室温下搅拌,tlc监测反应结束后,向反应液中加入乙酸乙酯,水洗、干燥,通过柱层析(乙酸乙酯/石油醚体系=1/1)分离纯化得到化合物d1。
[0071]
(4)二胺单体w-1的合成
[0072]
氮气气氛下,化合物d1(1.00g,1.70mmol)溶解在4ml水/乙醇(体积比=1/3)的混合液中,向混合液中加入还原zn粉(1.33g,20.39mmol)、氯化铵(363.51mg,6.80mmol)和冰乙酸(97.17mml,1.70mmol),50℃下搅拌,tlc监测反应结束后,趁热抽滤、萃取、干燥,将所得粗产物通过柱层析(乙酸乙酯/石油醚体系=2/1)分离纯化,获得化合物w-1。
[0073]
化合物w-1的质谱、核磁数据如图1-3所示。
[0074]
实施例2
[0075][0076]
合成方法参照实施例1,用代替合成化合物w-2。
[0077]
实施例3
[0078][0079]
合成方法参照实施例1,用代替合成化合物w-3。
[0080]
实施例4
[0081][0082]
化合物b、化合物c1的合成方法参照实施例1。
[0083]
(1)中间体d4的合成
[0084]
氮气气氛下,化合物c1(1.00g,2.20mmol)溶解于8ml干燥的丙酮中,向溶液中滴加三乙胺(672.99mml,4.84mmol),搅拌10min后,将丙烯酰氯(405.74mml,4.84mmol)分散到2ml干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,加入催化剂量的dmap催化反应进行,在室温下搅拌,tlc监测反应结束后,向反应液中加入乙酸乙酯,水洗、干燥,通过柱层析(乙酸乙酯/石油醚体系=1/1)分离纯化得到化合物d4。
[0085]
(2)二胺单体w-4的合成
[0086]
氮气气氛下,化合物d4(1.00g,1.78mmol)溶解在4ml水/乙醇(体积比=1/3)的混合液中,向混合液中加入还原zn粉(1.39g,21.33mmol)、氯化铵(380.37mg,7.11mmol)和冰乙酸(101.68mml,1.78mmol),50℃下搅拌,tlc监测反应结束后,趁热抽滤、萃取、干燥,将所得粗产物通过柱层析(乙酸乙酯/石油醚体系=2/1)分离纯化,获得化合物w-4。产物结构通过hrms鉴定。
[0087]
实施例5
[0088][0089]
合成方法参照实施例4,用代替合成化合物w-5。
[0090]
实施例6
[0091][0092]
合成方法参照实施例4,用代替合成化合物w-6。
[0093]
实施例7
[0094][0095]
合成方法参照实施例4,用代替合成化合物w-7。
[0096]
实施例8
[0097][0098]
合成方法参照实施例4,用代替合成化合物w-8。
[0099]
实施例9
[0100][0101]
合成方法参照实施例4,用代替合成化合物w-9。
[0102]
实施例10
[0103][0104]
(1)中间体b的合成
[0105]
氮气气氛中,将a(2.00g,11.10mmol)分散在5ml的去离子水中,将混合物加热到80℃,逐滴滴加浓hno3(65%,6.92ml,99.89mmol)/浓h2so4(98%,6.64ml,112.08mmol)的混合液,加热回流1h后,再次逐滴滴加浓hno3(65%,6.92ml,99.89mmol)/浓h2so4(98%,6.64ml,112.08mmol)的混合液,继续加热回流反应。tlc监测反应结束后,将反应液冷却,倒入冰水中,产生黄色沉淀,抽滤,滤饼洗涤3次后真空干燥,将干燥得到的粗产物在乙醇中重结晶,抽滤获得化合物b。
[0106]
(2)中间体c2的合成
[0107]
氮气气氛中,化合物b(2.00g,7.40mmol)和对苯二酚(2.04g,18.50mmol)混合均匀,将对甲苯磺酸(2.55g,14.80mmol)加入到混合物中,140℃下干粉反应,tlc监测反应结束后,降温,向反应瓶中加入15ml去离子水,搅拌0.5h,粗产物从反应混合物中沉淀出来,抽滤、洗涤、干燥后为棕色固体,将干燥后的粗产物通过柱层析(甲醇/二氯甲烷体系=1/50)分离提纯获得黄色固体化合物c2;
[0108]
(3)中间体d10的合成
[0109]
氮气气氛下,化合物c2(1.00g,2.20mmol)溶解于8ml干燥的丙酮中,向溶液中滴加三乙胺(672.99mml,4.84mmol),搅拌10min后,将甲基丙烯酰氯(468.62mml,4.84mmol)分散到2ml干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,加入催化剂量的dmap催化反应进行,在室温下搅拌,tlc监测反应结束后,向反应液中加入乙酸乙酯,水洗、干燥,通过柱层
析(乙酸乙酯/石油醚体系=1/1)分离纯化得到化合物d10。
[0110]
(4)二胺单体w-10的合成
[0111]
氮气气氛下,化合物d10(1.00g,1.70mmol)溶解在4ml水/乙醇(体积比=1/3)的混合液中,向混合液中加入还原zn粉(1.33g,20.39mmol)、氯化铵(363.51mg,6.80mmol)和冰乙酸(97.17mml,1.70mmol),50℃下搅拌,tlc监测反应结束后,趁热抽滤、萃取、干燥,将所得粗产物通过柱层析(乙酸乙酯/石油醚体系=2/1)分离纯化,获得化合物w-10。产物结构通过hrms鉴定。
[0112]
实施例11
[0113][0114]
合成方法参照实施例10,用代替合成化合物w-11。
[0115]
实施例12
[0116][0117]
合成方法参照实施例10,用代替合成化合物w-12。
[0118]
实施例13
[0119][0120]
化合物b、化合物c2合成方法参照实施例10。
[0121]
(1)中间体d13的合成
[0122]
氮气气氛下,化合物c2(1.00g,2.20mmol)溶解于8ml干燥的丙酮中,向溶液中滴加三乙胺(672.99mml,4.84mmol),搅拌10min后,将丙烯酰氯(405.74mml,4.84mmol)分散到2ml干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,加入催化剂量的dmap催化反应进行,在室温下搅拌,tlc监测反应结束后,向反应液中加入乙酸乙酯,水洗、干燥,通过柱层析(乙酸乙酯/石油醚体系=1/1)分离纯化得到化合物d13。
[0123]
(2)二胺单体w-13的合成
[0124]
氮气气氛下,化合物d13(1.00g,1.78mmol)溶解在4ml水/乙醇(体积比=1/3)的混合液中,向混合液中加入还原zn粉(1.39g,21.33mmol)、氯化铵(380.37mg,7.11mmol)和冰乙酸(101.68mml,1.78mmol),50℃下搅拌,tlc监测反应结束后,趁热抽滤、萃取、干燥,将所得粗产物通过柱层析(乙酸乙酯/石油醚体系=2/1)分离纯化,获得化合物w-13。产物结构通过hrms鉴定。
[0125]
实施例14
[0126][0127]
合成方法参照实施例13,用代替合成化合物w-14。
[0128]
实施例15
[0129][0130]
合成方法参照实施例13,用代替合成化合物w-15。
[0131]
实施例16
[0132][0133]
合成方法参照实施例13,用代替合成化合物w-16。
[0134]
实施例17
[0135][0136]
合成方法参照实施例13,用代替合成化合物w-17。
[0137]
实施例18
[0138][0139]
合成方法参照实施例13,用代替合成化合物w-18。
[0140]
实施例19
[0141][0142]
(1)中间体b的合成
[0143]
氮气气氛中,将a(2.00g,11.10mmol)分散在5ml的去离子水中,将混合物加热到80℃,逐滴滴加浓hno3(65%,6.92ml,99.89mmol)/浓h2so4(98%,6.64ml,112.08mmol)的混合液,加热回流1h后,再次逐滴滴加浓hno3(65%,6.92ml,99.89mmol)/浓h2so4(98%,6.64ml,112.08mmol)的混合液,继续加热回流反应。tlc监测反应结束后,将反应液冷却,倒入冰水中,产生黄色沉淀,抽滤,滤饼洗涤3次后真空干燥,将干燥得到的粗产物在乙醇中重结晶,抽滤获得化合物b。
[0144]
(2)中间体c3的合成
[0145]
氮气气氛中,化合物b(2.00g,7.40mmol)和邻苯二酚(2.04g,18.50mmol)混合均匀,将对甲苯磺酸(2.55g,14.80mmol)加入到混合物中,140℃下干粉反应,tlc监测反应结束后,降温,向反应瓶中加入15ml去离子水,搅拌0.5h,粗产物从反应混合物中沉淀出来,抽滤、洗涤、干燥后为棕色固体,将干燥后的粗产物通过柱层析(甲醇/二氯甲烷体系=1/50)分离提纯获得黄色固体化合物c3;
[0146]
(3)中间体d19的合成
[0147]
氮气气氛下,化合物c3(1.00g,2.20mmol)溶解于8ml干燥的丙酮中,向溶液中滴加三乙胺(672.99mml,4.84mmol),搅拌10min后,将甲基丙烯酰氯(468.62mml,4.84mmol)分散到2ml干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,加入催化剂量的dmap催化反应进行,在室温下搅拌,tlc监测反应结束后,向反应液中加入乙酸乙酯,水洗、干燥,通过柱层析(乙酸乙酯/石油醚体系=1/1)分离纯化得到化合物d19。
[0148]
(4)二胺单体w-19的合成
[0149]
氮气气氛下,化合物d19(1.00g,1.70mmol)溶解在4ml水/乙醇(体积比=1/3)的混合液中,向混合液中加入还原zn粉(1.33g,20.39mmol)、氯化铵(363.51mg,6.80mmol)和冰乙酸(97.17mml,1.70mmol),50℃下搅拌,tlc监测反应结束后,趁热抽滤、萃取、干燥,将所得粗产物通过柱层析(乙酸乙酯/石油醚体系=2/1)分离纯化,获得化合物w-19。产物结构通过hrms鉴定。
[0150]
实施例20
[0151][0152]
合成方法参照实施例19,用代替合成化合物w-20。
[0153]
实施例21
[0154][0155]
合成方法参照实施例19,用代替合成化合物w-21。
[0156]
实施例22
[0157][0158]
化合物b、化合物c3合成方法参照实施例19。
[0159]
(1)中间体d22的合成
[0160]
氮气气氛下,化合物c3(1.00g,2.20mmol)溶解于8ml干燥的丙酮中,向溶液中滴加三乙胺(672.99mml,4.84mmol),搅拌10min后,将丙烯酰氯(405.74mml,4.84mmol)分散到2ml干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,加入催化剂量的dmap催化反应进
行,在室温下搅拌,tlc监测反应结束后,向反应液中加入乙酸乙酯,水洗、干燥,通过柱层析(乙酸乙酯/石油醚体系=1/1)分离纯化得到化合物d22。
[0161]
(2)二胺单体w-22的合成
[0162]
氮气气氛下,化合物d22(1.00g,1.78mmol)溶解在4ml水/乙醇(体积比=1/3)的混合液中,向混合液中加入还原zn粉(1.39g,21.33mmol)、氯化铵(380.37mg,7.11mmol)和冰乙酸(101.68mml,1.78mmol),50℃下搅拌,tlc监测反应结束后,趁热抽滤、萃取、干燥,将所得粗产物通过柱层析(乙酸乙酯/石油醚体系=2/1)分离纯化,获得化合物w-22。产物结构通过hrms鉴定。
[0163]
实施例23
[0164][0165]
合成方法参照实施例23,用代替合成化合物w-23。
[0166]
实施例24
[0167][0168]
合成方法参照实施例23,用代替合成化合物w-24。
[0169]
实施例25
[0170][0171]
合成方法参照实施例23,用代替合成化合物w-25。
[0172]
实施例26
[0173][0174]
合成方法参照实施例23,用代替合成化合物w-26。
[0175]
实施例27
[0176][0177]
合成方法参照实施例23,用代替合成化合物w-27。
[0178]
实施例28
[0179]
可交联型光敏性二胺单体(w-28)的合成路线:
[0180][0181]
(1)中间体b的合成
[0182]
氮气气氛中,将a(2.00g,11.10mmol)分散在5ml的去离子水中,将混合物加热到80℃,逐滴滴加浓hno3(65%,6.92ml,99.89mmol)/浓h2so4(98%,6.64ml,112.08mmol)的混合液,加热回流1h后,再次逐滴滴加浓hno3(65%,6.92ml,99.89mmol)/浓h2so4(98%,6.64ml,112.08mmol)的混合液,继续加热回流反应。tlc监测反应结束后,将反应液冷却,倒入冰水中,产生黄色沉淀,抽滤,滤饼洗涤3次后真空干燥,将干燥得到的粗产物在乙醇中重结晶,抽滤获得化合物b。
[0183]
(2)中间体c4的合成
[0184]
氮气气氛中,化合物b(2.00g,7.40mmol)和间氨基苯酚(2.02g,18.50mmol)混合均匀,将对甲苯磺酸(2.55g,14.80mmol)加入到混合物中,140℃下干粉反应,tlc监测反应结束后,降温,向反应瓶中加入15ml去离子水,搅拌0.5h,粗产物从反应混合物中沉淀出来,抽滤、洗涤、干燥后为棕色固体,将干燥后的粗产物通过柱层析(甲醇/二氯甲烷体系=1/50)分离提纯获得棕色固体化合物c4;
[0185]
(3)中间体d28的合成
[0186]
氮气气氛下,化合物c4(1.00g,2.21mmol)溶解于8ml干燥的丙酮中,向溶液中滴加三乙胺(675.92mml,4.86mmol),搅拌10min后,将甲基丙烯酰氯(470.66mml,4.86mmol)分散到2ml干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,加入催化剂量的dmap催化反应进行,在室温下搅拌,tlc监测反应结束后,向反应液中加入乙酸乙酯,水洗、干燥,通过柱层析(乙酸乙酯/二氯甲烷体系=1/100)分离纯化得到化合物d28。
[0187]
(4)二胺单体w-31的合成
[0188]
氮气气氛下,化合物d28(1.00g,1.70mmol)溶解在4ml水/乙醇(体积比=1/3)的混合液中,向混合液中加入还原zn粉(1.33g,20.39mmol)、氯化铵(363.51mg,6.80mmol)和冰乙酸(97.17mml,1.70mmol),50℃下搅拌,tlc监测反应结束后,趁热抽滤、萃取、干燥,将所得粗产物通过柱层析(甲醇/二氯甲烷体系=1/60)分离纯化,获得化合物w-28。产物结构通过hrms鉴定。
[0189]
实施例29
[0190][0191]
合成方法参照实施例28,用代替合成化合物w-29。
[0192]
实施例30
[0193][0194]
合成方法参照实施例28,用代替合成化合物w-30。
[0195]
实施例31
[0196][0197]
化合物b、化合物c4合成方法参照实施例28。
[0198]
(1)中间体d31的合成
[0199]
氮气气氛下,化合物c4(1.00g,2.21mmol)溶解于8ml干燥的丙酮中,向溶液中滴加三乙胺(675.92mml,4.86mmol),搅拌10min后,将丙烯酰氯(407.50mml,4.86mmol)分散到2ml干燥的丙酮中,冰浴下逐滴滴加到上述反应混合液中,加入催化剂量的dmap催化反应进行,在室温下搅拌,tlc监测反应结束后,向反应液中加入乙酸乙酯,水洗、干燥,通过柱层析(乙酸乙酯/二氯甲烷体系=1/100)分离纯化得到化合物d31。
[0200]
(4)二胺单体w-31的合成
[0201]
氮气气氛下,化合物d31(1.00g,1.78mmol)溶解在4ml水/乙醇(体积比=1/3)的混合液中,向混合液中加入还原zn粉(1.40g,21.41mmol)、氯化铵(381.71mg,7.14mmol)和冰
乙酸(102.03mml,1.78mmol),50℃下搅拌,tlc监测反应结束后,趁热抽滤、萃取、干燥,将所得粗产物通过柱层析(甲醇/二氯甲烷体系=1/60)分离纯化,获得化合物w-31。产物结构通过hrms鉴定。
[0202]
实施例32
[0203][0204]
合成方法参照实施例31,用代替合成化合物w-32。
[0205]
实施例33
[0206][0207]
合成方法参照实施例31,用代替合成化合物w-33。
[0208]
实施例34
[0209][0210]
合成方法参照实施例31,用代替合成化合物w-34。
[0211]
实施例35
[0212][0213]
合成方法参照实施例31,用代替合成化合物w-35。
[0214]
实施例36
[0215][0216]
合成方法参照实施例31,用代替合成化合物w-36。
[0217]
实施例37
[0218][0219]
化合物w-1的合成方法参照实施例1。
[0220]
可光交联型光敏聚酰亚胺(pspi-1)的合成路线
[0221]
在惰性气体气氛下,将化合物w-1(100.00mg,188.47μmol)溶解于干燥且除氧的2ml氮甲基吡咯烷酮中,待完全溶解后,向溶液中加入3,3’,4,4
’‑
二苯甲酮四甲酸二酐(63.77mg,197.90μmol),25℃下搅拌反应8h,将反应液滴加到乙酸乙酯中,有黄色物质沉出、洗涤、干燥,获得聚酰胺酸paa-1;向聚酰胺酸paa-1在氮甲基吡咯烷酮的溶液中加入0.5ml三乙胺、0.5ml吡啶、2ml甲苯,180℃下脱水环化5h,降温,将反应液滴加到无水乙醇中沉出、洗涤、干燥,获得可光交联型光敏聚酰亚胺pspi-1。
[0222]
paa-1与pspi-1的傅里叶红外光谱数据如图4所示。对比paa-1的曲线,pspi-1曲线中可以看出,1363cm-1
处为酰亚胺环上c-n键的伸缩振动吸收峰,1783cm-1
与1731cm-1
处分别为酰亚胺环上2个c=o的对称与不对称伸缩振动吸收峰,可以确定酰亚胺环结构的存在;单
体w-1结构中具有甲基丙烯酸酯基团,在pspi-1图谱中可以明确观察到,1608cm-1
处为甲基丙烯酸酯结构中c=c的特征吸收峰,由此可确定制备了pspi-1。
[0223]
实施例38
[0224][0225]
合成方法参照实施例37,合成聚合物paa-2、pspi-2。
[0226]
实施例39
[0227][0228]
合成方法参照实施例37,合成聚合物paa-3、pspi-3。
[0229]
实施例40
[0230][0231]
合成方法参照实施例37,合成聚合物paa-4、pspi-4。
[0232]
实施例41
[0233]
[0234]
化合物w-7的合成方法参照实施例7。
[0235]
可光交联型光敏聚酰亚胺(pspi-5)的合成路线
[0236]
在惰性气体气氛下,将化合物w-7(100.00mg,152.74μmol)溶解于干燥且除氧的2ml氮甲基吡咯烷酮中,待完全溶解后,向溶液中加入2,2-二(3,4-二羧基苯基)甲烷二酐(49.43mg,160.37μmol),25℃下搅拌反应8h,将反应液滴加到乙酸乙酯中,有黄色物质沉出、洗涤、干燥,获得聚酰胺酸paa-5;向聚酰胺酸paa-5在氮甲基吡咯烷酮的溶液中加入0.5ml三乙胺、0.5ml吡啶、2ml甲苯,180℃下脱水环化5h,降温,将反应液滴加到无水乙醇中沉出、洗涤、干燥,获得可光交联型光敏聚酰亚胺pspi-5。
[0237]
实施例42
[0238][0239]
合成方法参照实施例41,合成聚合物paa-6、pspi-6。
[0240]
实施例43
[0241][0242]
合成方法参照实施例41,合成聚合物paa-7、pspi-7。
[0243]
实施例44
[0244][0245]
化合物w-31的合成方法参照实施例31。
[0246]
可光交联型光敏聚酰亚胺(pspi-8)的合成路线
[0247]
在惰性气体气氛下,将化合物w-31(100.00mg,199.78μmol)溶解于干燥且除氧的2ml氮甲基吡咯烷酮中,待完全溶解后,向溶液中加入二苯并二氧六环二酐(68.01mg,209.77μmol),25℃下搅拌反应8h,将反应液滴加到乙酸乙酯中,有黄色物质沉出、洗涤、干燥,获得聚酰胺酸paa-8;向聚酰胺酸paa-8在氮甲基吡咯烷酮的溶液中加入0.5ml三乙胺、
0.5ml吡啶、2ml甲苯,180℃下脱水环化5h,降温,将反应液滴加到无水乙醇中沉出、洗涤、干燥,获得可光交联型光敏聚酰亚胺pspi-8。
[0248]
实施例45
[0249][0250]
合成方法参照实施例44,合成聚合物paa-9、pspi-9。
[0251]
实施例46
[0252][0253]
合成方法参照实施例44,合成聚合物paa-10、pspi-10。
[0254]
实施例47
[0255][0256]
合成方法参照实施例44,合成聚合物paa-11、pspi-11。
[0257]
实施例48
[0258]
[0259]
合成方法参照实施例44,合成聚合物paa-12、pspi-12。
[0260]
实施例49
[0261]
将30mg pspi-1溶解在0.2ml干燥的dmac中,加入5%含量的二苯基-(2,4,6-三甲基苯甲酰)氧磷(光引发剂tpo),在室温下搅拌2h,在洁净的玻璃片上涂膜,干燥除去溶剂后,390nm紫外光照射后取样品做红外光谱测试(nicolet in10显微红外光谱仪)。图5为可光交联聚酰亚胺pspi-1紫外光照射前后的红外光谱图,从图中可以看出,与紫外光照射前pspi-1相比,光交联后的cl-pspi-1在1606cm-1
位置处c=c双键的红外特征吸收峰强度明显减弱,证明紫外光照射下发生了光交联反应。
[0262]
即表明聚酰亚胺pspi-1可以在紫外照射下发生光交联。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1