一种生产L-精氨酸的基因工程菌及其应用
glutamicum atcc 13032的精氨酸生物合成相关基因簇argc、argj、argb、argd、argf、argg、argh以及编码精氨酸转运蛋白的基因lyse;并且异源过表达来源于b.subtilis a260的编码氨甲酰磷酸合成酶的基因pyraa、pyrab;并且含有氨基酸序列如seq id no.1所示的大肠杆菌rna聚合酶β-亚基rpob突变体。
9.第二方面,本发明提供了如上所述的基因工程菌的构建方法,包括:在宿主大肠杆菌中引入corynebacterium glutamicum atcc 13032的精氨酸生物合成相关基因簇argc、argj、argb、argd、argf、argg、argh以及编码精氨酸转运蛋白的基因lyse并过表达;引入b.subtilis a260的编码氨甲酰磷酸合成酶的基因pyraa、pyrab并过表达;并且将以下基因敲除或失活:编码精氨酸脱羧酶的基因spea、编码精氨酸脱羧酶的基因adia和编码精氨酸琥珀酰转移酶的基因asta;并将大肠杆菌自身rna聚合酶β-亚基rpob的第564位氨基酸由脯氨酸突变为苏氨酸。
10.第三方面,本发明提供了如上所述的基因工程菌在高效生产l-精氨酸中的应用。
11.第四方面,本发明提供了利用如上所述的基因工程菌生产l-精氨酸的方法,包括:在培养基中培养所述基因工程菌以使其生产l-精氨酸;以及,从所述基因工程菌和/或所述培养基中收集所述l-精氨酸。
12.第五方面,本发明提供一种大肠杆菌rna聚合酶β-亚基rpob突变体,其氨基酸序列如seq id no.1所示。
13.本发明的有益效果如下:
14.本发明选用生长周期短、代谢途径清晰、分子操作便捷的大肠杆菌为出发菌株,从l-精氨酸合成代谢途径的基因工程改造和整个代谢网络的工程改造出发,对大肠杆菌中精氨酸合成途径和整个氨基酸代谢网络中与精氨酸相关的代谢流进行分析重构,得到一株遗传背景清晰,不携带质粒,不经诱变且能稳定高效生产l-精氨酸的基因工程菌株。
15.rpob基因编码rna聚合酶β-亚基,与细胞内全局转录调控有关,对维持细胞代谢平衡及碳源分配具有重要作用,但该基因与l-精氨酸合成途径的关系及作用尚不清晰。本发明证实,rpob p564t突变体可以显著提升大肠杆菌生产l-精氨酸的生产水平。具体而言,与出发菌株arg9相比,含有rpob p564t突变体的基因工程菌株arg10中l-精氨酸积累浓度从24.5g/l提升到30.1g/l,l-精氨酸产率提高了22.9%。
具体实施方式
16.下面通过具体的实施方案叙述本发明。除非特别说明,本发明中所用的技术手段均为本领域技术人员所公知的方法。另外,实施方案应理解为说明性的,而非限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。对于本领域技术人员而言,在不背离本发明实质和范围的前提下,对这些实施方案中的物料成分和用量进行的各种改变或改动也属于本发明的保护范围。
17.第一方面,本发明提供了一种生产l-精氨酸的基因工程菌,该基因工程菌以大肠杆菌为宿主,不表达以下基因:编码精氨酸脱羧酶的基因spea、编码精氨酸脱羧酶的基因adia和编码精氨酸琥珀酰转移酶的基因asta;并且异源过表达来源于corynebacterium glutamicum atcc 13032的精氨酸生物合成相关基因簇argc、argj、argb、argd、argf、argg、argh以及编码精氨酸转运蛋白的基因lyse;并且异源过表达来源于b.subtilis a260的编
码氨甲酰磷酸合成酶的基因pyraa、pyrab;并且含有氨基酸序列如seq id no.1所示的大肠杆菌rna聚合酶β-亚基rpob突变体。
18.根据本发明,用于构建所述基因工程菌的宿主可以是任意的大肠杆菌,例如,本领域常见用作基因工程宿主的e coli mg1655、e coli w3110、e coli bl21、e.coli bw25113等模式菌株,只要可以对其进行基因工程改造表达上述需要过表达或异源过表达的基因即可;上述大肠杆菌各基因的选择根据所选宿主进行选择,但均是实现同一功能的基因。
19.根据本发明一种优选的实施方式,所述宿主是e.coli mg1655。
20.根据本发明,不表达上述基因的方式可以采取本领域常规手段,例如,通过本领域常规手段使该基因失活或者将该基因敲除。
21.根据本发明,所述不表达是指该基因表达产物的量显著低于原有水平,例如,显著降低了至少50%、60%、70%、80%、90%、100%。
22.根据本发明,所述过表达是指该基因表达产物的量显著高于原有水平,例如,提高了150%或更多,200%或更多,300%或更多。
23.根据本发明,过表达上述基因的方式,可以通过在大肠杆菌基因组中引入和/或增加该基因的拷贝数(例如,通过pet28a、ptrc99a、pstv28等自主复制质粒增加基因的拷贝数,或者增加大肠杆菌染色体中该基因的拷贝数),或者修饰该基因的表达调控序列(例如,启动子,核糖体结合位点等),或者以上方法的组合。
24.根据本发明一种优选的实施方式,过表达上述基因的方式是将该基因连接了强启动子并整合在大肠杆菌基因组中,其中基因的整合位点可以根据本领域技术人员常规认知选择一些不会对细菌生长和基础代谢产生明显影响的假基因位点,例如yeep、ygip、yghx、ygay、yjit、yjip、ycjv、ycgh、ygay、yeel、ilvg、rph等基因位点都是可以选择的;所述强启动子可以选择p
trc
、bba-j23100、t7中任意一种,优选p
trc
。
25.第二方面,本发明提供了如上所述的基因工程菌的构建方法,包括:在宿主大肠杆菌中引入corynebacterium glutamicum atcc 13032的精氨酸生物合成相关基因簇argc、argj、argb、argd、argf、argg、argh以及编码精氨酸转运蛋白的基因lyse并过表达;引入b.subtilis a260的编码氨甲酰磷酸合成酶的基因pyraa、pyrab并过表达;并且将以下基因敲除或失活:编码精氨酸脱羧酶的基因spea、编码精氨酸脱羧酶的基因adia和编码精氨酸琥珀酰转移酶的基因asta;并将大肠杆菌自身rna聚合酶β-亚基rpob的第564位氨基酸由脯氨酸突变为苏氨酸。
26.本发明第一方面已经详细介绍了上述各基因的选择、宿主的选择等,此处不再赘述第一方面已经介绍过的内容。
27.根据本发明一种具体的实施方式,该方法包括:
28.(1)阻断精氨酸降解途径:
29.以大肠杆菌为宿主,敲除其基因组中编码精氨酸脱羧酶的基因spea、编码精氨酸脱羧酶的基因adia和编码精氨酸琥珀酰转移酶的基因asta;
30.(2)强化精氨酸合成途径:
31.将来源于corynebacterium glutamicum atcc 13032的精氨酸生物合成相关基因簇argc、argj、argb、argd、argf、argg、argh整合在宿主大肠杆菌基因组中,并由启动子p
trc
控制其表达;强化精氨酸合成途径代谢流量,进而使更多的碳源流向精氨酸合成方向,促进
胞内精氨酸的积累;
32.(3)提高精氨酸合成过程中前体物氨甲酰磷酸供应:
33.将来源于b.subtilis a260的编码氨甲酰磷酸合成酶的基因pyraa、pyrab整合在宿主大肠杆菌基因组中,并由启动子p
trc
控制其表达;强化氨甲酰磷酸合成酶,以提高前体物质氨甲酰磷酸的供应;
34.(4)增强胞内精氨酸向胞外转移的能力:
35.将来源于corynebacterium glutamicum atcc 13032的编码精氨酸转运蛋白的基因lyse整合在宿主大肠杆菌基因组中,并由启动子p
trc
控制其表达;以加强胞内精氨酸向胞外转运的能力;
36.(5)整合rpob p564t突变体:
37.将宿主大肠杆菌自身编码rna聚合酶β-亚基的基因rpob第1690位单核苷酸碱基由c突变为a;形成rpob p564t蛋白突变体,促进大肠杆菌中l-精氨酸合成。
38.第三方面,本发明提供了如上所述的基因工程菌在高效生产l-精氨酸中的应用。
39.第四方面,本发明提供了利用如上所述的基因工程菌生产l-精氨酸的方法,包括:在培养基中培养所述基因工程菌以使其生产l-精氨酸;以及,从所述基因工程菌和/或所述培养基中收集所述l-精氨酸。
40.根据本发明第四方面,所述基因工程菌的培养可以采用本领域常规方法进行。用于生产麦角硫因的培养基可以是合成的或天然的培养基,例如含有碳源、氮源、硫源、无机离子和需要的其它有机和无机组分的典型培养基。
41.所述基因工程菌可以在需氧条件下进行培养,持续16至72小时,或20至60小时,或26至48小时;培养温度可以控制在30至45℃,或30至37℃内;且ph可以调节在5.0至8.0之间,或6.0至7.5之间,或6.8至7.2之间。可以通过使用无机或有机酸性或碱性物质,以及氨气调节ph。
42.培养后,可以通过常规技术(例如离心,膜过滤)从液体培养基中除去固体,如细胞和细胞碎片,然后可以通过常规技术(例如浓缩,离子交换层析,结晶)的任意组合从发酵液回收麦角硫因。
43.根据本发明一种优选的实施方式,优选的发酵培养基组成为:葡萄糖20-40g/l,酵母提取物1-3g/l,蛋白胨2-3g/l,k2hpo
4 3-6g/l,mgso4·
7h2o 1-2g/l,feso4·
7h2o 15-20mg/l,mnso4·
7h2o 15-20mg/l,vb1、vb3、vb5、vb12、vh各1-3mg/l,其余为水,ph 7.0-7.2。
44.第五方面,本发明提供一种大肠杆菌rna聚合酶β-亚基突变体rpob p564t,其氨基酸序列如seq id no.1所示。根据本发明,所述突变体相比亲本的rpob具有提高大肠杆菌l-精氨酸合成水平的效果。优选的,编码所述突变体rpob p564t的基因的核苷酸序列如seq id no.2所示。
45.以下将通过具体的实施例对本发明进行更详细描述。以下实施例中:
46.如无特别说明,本发明实施例中所涉及的基因编辑方法参照文献(li y,lin z,huang c,et al.metabolic engineering of escherichia coli using crispr
–
cas9 meditated genome editing.metabolic engineering,2015)进行。其中predcas9携带grna表达质粒pgrb的消除系统,λ噬菌体的red重组系统及cas9蛋白表达系统,奇霉素抗性(工作浓度:100mg/l),32℃培养;pgrb以puc18为骨架,包括启动子j23100,grna-cas9结合区域序
列和终止子序列,氨苄青霉素抗性(工作浓度:100mg/l),37℃培养。该方法通过pgrb质粒和重组dna片段的转化,筛选阳性菌株后再将质粒消除,实现重组片段的敲除或敲入。其中用于敲除的重组片段由需敲除基因的上下游同源臂组成(上游同源臂-下游同源臂),用于整合的重组片段以整合位点的上下游同源臂及待整合的基因片段组成(上游同源臂-目的基因-下游同源臂)。其他所涉及的分子生物学、基因工程等具体操作手段根据本领域人员容易获得的技术手册、教科书或文献报道均可以实现,不必在此详细描述操作过程。此外,以下实施例中选用了具体的菌株作为宿主,因而根据宿主选择了具体的基因整合位点、目标基因及其引物等,但并不意味着本发明目的仅能通过这些具体的选择得以实现,不能以此限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。
47.实施例1:大肠杆菌基因工程菌arg10的构建
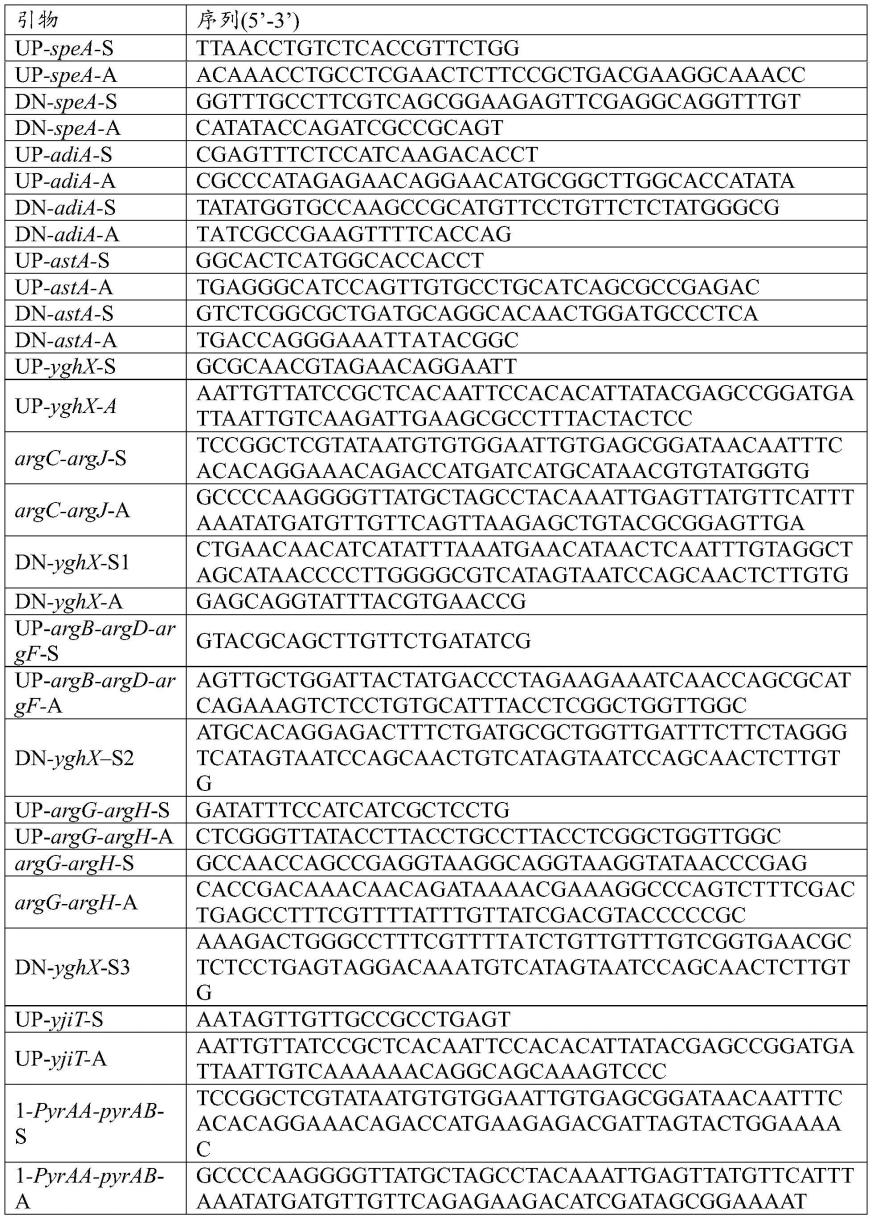
48.1菌株构建过程中用到的引物如下表所示:
49.50.[0051][0052]
2菌株的构建过程
[0053]
2.1敲除spea基因
[0054]
以e.coli mg1655基因组为模板,根据其spea基因(ncbi-geneid:947432)的上下游序列设计上游同源臂引物(up-spea-s、up-spea-a)和下游同源臂引物(dn-spea-s、dn-spea-a),并pcr扩增其上下游同源臂片段。上述片段通过重叠pcr的方法融合获得spea基因的敲除片段(上游同源臂-下游同源臂)。将引物grna-spea-s和grna-spea-a退火后得到的dna片段与质粒pgrb连接,构建重组质粒pgrb-spea。制备e.coli mg1655的感受态细胞,将质粒pgrb-spea和spea敲除片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg1。阳性菌株通过pcr及电泳验证spea基因敲除成功。
[0055]
2.2敲除adia基因
[0056]
以e.coli mg1655基因组为模板,根据其adia基因(ncbi-geneid:948638)的上下游序列设计上游同源臂引物(up-adia-s、up-adia-a)和下游同源臂引物(dn-adia-s、dn-adia-a),并pcr扩增其上下游同源臂片段。上述片段通过重叠pcr的方法融合获得adia基因的敲除片段(上游同源臂-下游同源臂)。将引物grna-adia-s和grna-adia-a退火得到的dna片段与质粒pgrb连接,构建pgrb-adia。制备arg1的感受态细胞,将质粒pgrb-adia和adia敲除片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg2。阳性菌株通过pcr及电泳验证adia基因敲除成功。
[0057]
2.3敲除asta基因
[0058]
以e.coli mg1655基因组为模板,根据其asta基因(ncbi-geneid:946261)的上下游序列设计上游同源臂引物(up-asta-s、up-asta-a)和下游同源臂引物(dn-asta-s、dn-asta-a),并pcr扩增其上下游同源臂片段。上述片段通过重叠pcr的方法融合获得asta基因的敲除片段(上游同源臂-下游同源臂)。将引物grna-asta-s和grna-asta-a的退火制得的dna片段与质粒pgrb连接,构建pgrb-asta。制备arg2的感受态细胞,将质粒pgrb-asta和asta敲除片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg3。阳性菌株通过pcr及电泳验证asta基因敲除成功。
[0059]
2.4整合corynebacterium glutamicum atcc 13032的精氨酸生物合成相关基因簇
[0060]
将corynebacterium glutamicum atcc 13032的精氨酸生物合成相关基因簇(包含argc、argj、argb、argd、argf、argg、argh七个基因)按顺序依次整合至大肠杆菌yghx基因位点,并由启动子p
trc
控制该外源基因簇的转录表达,构建了菌株arg6。以下分三段整合:
[0061]
2.4.1p
trc-argc-argj的整合
[0062]
以e.coli mg1655基因组为模板,根据其yghx基因的上下游序列设计上游同源臂引物(up-yghx-s、up-yghx-a)和下游同源臂引物(dn-yghx-s1、dn-yghx-a),pcr扩增其上下游同源臂片段;以谷氨酸棒状杆菌(atcc 13032)基因组为模板,根据其argc-argj基因序列(ncbi-geneid:1019370、1019371)设计引物(argc-argj-s、argc-argj-a),pcr扩增argc-argj片段;启动子p
trc
则设计在上游同源臂的下游引物和argc-argj基因的上游引物中。上述片段通过重叠pcr的方法融合,获得argc-argj基因的整合片段(上游同源臂-p
trc-argc-argj-下游同源臂),将引物grna-yghx-s和grna-yghx-a退火制得含靶序列的dna序列,与质粒pgrb连接,构建pgrb-yghx。制备arg3的感受态细胞,将质粒pgrb-yghx和argc-argj基因的整合片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg4。阳性菌株通过pcr及电泳验证p
trc-argc-argj片段整合成功。
[0063]
2.4.2argb-argd-argf的整合
[0064]
以谷氨酸棒状杆菌(atcc13032)基因组为模板,根据argb-argd-argf(ncbi-geneid:1019372、1019373、1019374)及其上游序列设计上游同源臂引物(up-argb-argd-argf-s、up-argb-argd-argf-a),pcr扩增其上游同源臂片段;以e.coli mg1655基因组为模板,根据其yghx基因的下游序列设计下游同源臂引物(dn-yghx-s2、dn-yghx-a),pcr扩增其下游同源臂片段。上述片段通过重叠pcr的方法融合,获得argb-argd-argf的整合片段(argb的上游片段-argb-argd-argf-下游同源臂)。将引物grna-argbdf-s和grna-argbdf-a退火制得含靶序列的dna片段,与质粒pgrb连接,构建pgrb-argbdf。制备arg4的感受态细胞,将质粒pgrb-argbdf和argb-argd-argf的整合片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg5。阳性菌株通过pcr及电泳验证argb-argd-argf片段整合成功。
[0065]
2.4.3argg-argh的整合
[0066]
以谷氨酸棒状杆菌(atcc13032)基因组为模板,根据argg-argh(ncbi-geneid:1019376、1019377)及其上游序列设计上游同源臂引物(up-argg-argh-s、up-argg-argh-a)及argg-argh片段引物(argg-argh-s、argg-argh-a),pcr扩增其上游同源臂片段和argg-argh片段;以e.coli mg1655基因组为模板,根据其yghx基因的下游序列设计下游同源臂引物(dn-yghx-s3、dn-yghx-a),pcr扩增其下游同源臂片段。上述片段通过重叠pcr的方法融合,获得argg-argh的整合片段(argg的上游片段-argg-argh-下游同源臂)。将引物grna-argg-argh-s和grna-argg-argh-a退化制得制得含靶序列的dna序列,与质粒pgrb连接,构建pgrb-argg-argh。制备arg5的感受态细胞,将质粒pgrb-argg-argh和argg-argh的整合片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg6。阳性菌株通过pcr及电泳验证argg-argh片段整合成功。
[0067]
2.5整合b.subtilis a260的pyraa-pyrab基因
[0068]
b.subtilis a260是以枯草芽孢杆菌168菌株为出发菌株,采用artp诱变和高通量筛选相结合的方法选育而来(记载于cn105671007a,保藏编号:cgmcc no.11775)。该菌株解
除了尿苷酸及精氨酸对氨甲酰磷酸合成酶的反馈调节作用,通过对其嘧啶核苷操纵子基因进行测序发现其pyrab编码的氨甲酰磷酸大亚基第949位的谷氨酸缺失(突变体氨基酸序列记载于cn105671007a)。将b.subtilis a260中不受精氨酸反馈抑制的氨甲酰磷酸合成酶(pyraa、pyrab)整合至大肠杆菌中,以提高精氨酸合成过程中前体物氨甲酰磷酸的供应量。
[0069]
枯草芽孢杆菌a260中pyraa-pyrab基因共4308bp分两段整合至大肠杆菌中,其中第一段长度为2667bp,第二段长度为1641bp。
[0070]
2.5.1第一段p
trc-pyraa-pyrab的整合
[0071]
以e.coli mg1655基因组为模板,根据其yjit基因的上下游序列设计上游同源臂引物(up-yjit-s、up-yjit-a)和下游同源臂引物(dn-yjit-s、dn-yjit-a),pcr扩增其上下游同源臂片段;以b.subtilis a260(cgmcc no.11775)基因组为模板,根据基因pyraa(ncbi-geneid:937368)、pyrab(ncbi-geneid:936608)设计引物(1-pyraa-pyrab-s、1-pyraa-pyrab-a),扩增第一段pyraa-pyrab基因片段。启动子p
trc
则设计在上游同源臂的下游引物和pyraa-pyrab基因的上游引物中。上述片段通过重叠pcr的方法融合获得第一段pyraa-pyrab基因的整合片段(上游同源臂-p
trc-pyraa-pyrab-下游同源臂),将引物grna-yjit-s和grna-yjit-a退火制得含靶序列的dna片段,与质粒pgrb连接,构建pgrb-yjit。制备arg6的感受态细胞,将质粒pgrb-yjit和第一段pyraa-pyrab基因的整合片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg7。阳性菌株通过pcr及电泳验证第一段p
trc-pyraa-pyrab片段整合成功。
[0072]
2.5.2第二段pyraa-pyrab的整合
[0073]
以b.subtilis a260(cgmcc no.11775)基因组为模板,根据第二段pyraa-pyrab基因序列及其上游序列设计上游同源臂引物(2-pyraa-pyrab-s、2-pyraa-pyrab-a),pcr扩增其上游同源臂片段(包括第一段pyraa-pyrab下游序列266bp和整合第二段pyraa-pyrab序列1641bp共1907bp);以e.coli mg1655基因组为模板,根据其yjit基因的下游序列设计下游同源臂引物(dn-yjit-s1、dn-yjit-a),pcr扩增其下游同源臂片段。上述片段通过重叠pcr的方法融合,获得第二段pyraa-pyrab的整合片段(第二段pyraa-pyrab-下游同源臂)。将引物grna-pyraa-pyrab-s和grna-pyraa-pyrab-a退火制得含靶序列的dna片段,与质粒pgrb连接,构建pgrb-pyraa-pyrab。制备arg7的感受态细胞,将质粒pgrb-pyraa-pyrab和第二段pyraa-pyrab基因的整合片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg8。阳性菌株通过pcr及电泳验证第二段pyraa-pyrab片段整合成功。
[0074]
2.6整合corynebacterium glutamicum atcc 13032的lyse基因
[0075]
以e.coli mg1655基因组为模板,根据其trpr基因的上下游序列设计上游同源臂引物(up-trpr-s、up-trpr-a)和下游同源臂引物(dn-trpr-s、dn-trpr-a),pcr扩增其上下游同源臂片段;根据lyse基因(ncbi-geneid:1019244)设计引物(lyse-s、lyse-a),扩增lyse基因片段。启动子p
trc
则设计在上游同源臂的下游引物和lyse基因的上游引物中。上述片段通过重叠pcr的方法融合获得lyse基因的整合片段(上游同源臂-p
trc-lyse-下游同源臂),构建pgrb-trpr使用的含靶序列的dna片段通过引物grna-trpr-s和grna-trpr-a的退火制得。制备arg8的感受态细胞,将质粒pgrb-trpr和lyse基因的整合片段同时电转化入感受态细胞,筛选阳性菌株后再将质粒消除获得菌株arg9。阳性菌株通过pcr及电泳验证p
trc-lyse片段整合成功。
[0076]
2.7整合rpob p564t突变体
[0077]
以e.coli mg1655基因组为模板,根据其rpob基因(ncbi-geneid:948488)的上下游序列设计上游同源臂引物(up-rpob-s、up-rpob-a)和下游同源臂引物(dn-rpob-s、dn-rpob-a),其中dn-rpob-a引物包含rpob基因第1690处单核苷酸碱基由c突变为a。利用上述引物pcr扩增上下游同源臂,其中下游同源臂包含rpob氨基酸序列第564处脯氨酸突变为苏氨酸。上述片段通过重叠pcr的方法融合获得rpob p564t基因的整合片段(上游同源臂-下游同源臂);构建pgrb-rpob使用的含靶序列的dna片段通过引物grna-rpob-s和grna-rpob-a的退火制得。制备arg9的感受态细胞,将质粒pgrb-rpob和rpob p564t整合片段同时电转化入arg9感受态细胞,筛选整合rpob p564t的阳性菌株并消除质粒获得菌株arg10。阳性菌株通过pcr及电泳验证rpob p564t片段整合成功。
[0078]
实施例2:基因工程菌摇瓶发酵生产l-精氨酸
[0079]
利用上述构建的基因工程菌株(arg9、arg10)摇瓶发酵生产l-精氨酸的方法如下:
[0080]
斜面培养:取-80℃保藏菌种划线接种于活化斜面,37℃培养12h,并传代一次;
[0081]
摇瓶种子培养:用接种环刮取一环斜面种子接种于装有30ml种子培养基的500ml三角瓶中,九层纱布封口,37℃,200rpm培养7-10h;
[0082]
摇瓶发酵培养:按种子培养液体积10-15%的接种量接种到装有发酵培养基的500ml三角瓶中(终体积为30ml),九层纱布封口,37℃,200r/min振荡培养,发酵过程中通过补加氨水维持ph在7.0-7.2;补加60%(m/v)葡萄糖溶液维持发酵进行;
[0083]
培养基的组成:(1)斜面培养:葡萄糖1g/l、蛋白胨10g/l、牛肉膏10g/l、酵母粉5g/l、nacl 2.5g/l、琼脂粉25g/l。(2)摇瓶种子培养及发酵培养:葡萄糖20g/l,酵母提取物3g/l,蛋白胨2g/l,k2hpo
4 6g/l,mgso4·
7h2o 1g/l,feso4·
7h2o 20mg/l,mnso4·
7h2o 20mg/l,v
b1
、v
b3
、v
b5
、v
b12
、vh各1mg/l其余为水,ph 7.0-7.2。
[0084]
经摇瓶28h发酵培养后,各取1ml发酵液13000转/分钟高速去除菌体,获取发酵上清液。利用高效液相色谱检测发酵上清液中l-精氨酸浓度(如表1所示)。
[0085]
表1菌株摇瓶发酵结果
[0086][0087]
表1结果显示,与野生型e.coli mg1655相比,arg9中l-精氨酸浓度从无提升到24.5g/l。以上实验结果表明,通过逐步加强精氨酸合成代谢网络,可以显著提高了e.coli mg1655中l-精氨酸生产性能。与arg9相比,含有rpob p564t突变体基因的菌株arg10将l-精氨酸产量进一步从24.5g/l提升到30.1g/l,提升了22.9%。以上实验结果表明,rpob p564t突变体相比亲本的rna聚合酶β-亚基具有提高大肠杆菌中l-精氨酸合成水平的效果,显著提高了基因工程菌对于l-精氨酸的生产能力。
[0088]
虽然本发明已经以较佳实施例公开如上,但其并非用以限定本发明,任何本领域技术人员,在不脱离本发明的精神和原理的情况下,可以对这些实施例进行各种形式和细
节的变化、修改、替换和变型,本发明的范围由权利要求及其等同物所限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1