一种苝醌染料及其制备方法与流程
本发明涉及化合物及其制备领域,具体涉及一种具有苝醌母核的化合物的制备方法。
背景技术:
苝醌类衍生物(pqd)是一类广泛分布于自然界的天然色素,是一种潜在的优良光敏剂,传统上被用于治疗风湿性关节炎和胃病;最近研究发现,pqd可以作为新一代的光动力抗肿瘤药物,另外,还有望发展成为新型的抗病毒药物以及广谱抗生素。苝醌类类衍生物具有光增强活性,拥有广阔的应用前景。
为了实现pqd相关技术的产业化,必须解决pqd的制备问题。目前,由于qpd的化学合成还比较缺乏,现有的制备方法大多通过真菌发酵方式。
例如,公开号为cn105624226a的中国专利文献公开了一种苝醌化合物的生物合成方法,采用子囊菌纲,肉座菌科,菌寄生菌属的真菌为菌种进行发酵制得痂囊腔菌素、竹红菌甲素、竹红菌乙素、寄生菌菌素a和b等苝醌类化合物。
公开号为cn102302089a的中国专利文献也公开了一种采用复杂菌种体系发酵的制备方法。公开号为cn1680562a的中国专利文献公开了一种采用子囊菌纲,肉座菌科,菌寄生菌属的真菌ascomycetesypomyces(fr)tul.sp为菌种发酵制得苝醌化合物产品的方法。
再如,公开号为cn103642864a的中国专利文献公开了一种利用内生竹黄真菌发酵制备苝醌类化合物的方法。公开号为cn102219768a的中国专利文献也公开了一种由蜈蚣衣属植物地衣的内生真菌发酵液中提取分离得到的苝醌类化合物的方法。
现有的苝醌类化合物大多采用生物发酵或者直接从天然原材料中提取,制备方法成本高、效率低、污染大。更重要的是,苝醌溶解性差且不具有选择性功能位点,难以修饰,这很大程度上限制了苝醌类染料的发展与应用。
技术实现要素:
为克服现有苝醌染料制备方法成本高、效率低、结构修饰难度大等技术问题,本发明公开了一种苝醌染料的制备方法,旨在提供一种苝母核化学合成、修饰方法。
一种苝醌染料的制备方法,式1的原料与氧化剂在有机溶剂的水溶液中氧化偶联,再经酸析、结晶制得所述的苝醌染料;
式1中,所述的r1、r2独自选自h、-cl、-br、-i、-cn、c1-c24烷基、c3-c24环烷基、c1-c24烷氧基、卤代c1-c24烷基、卤代c3-c24环烷基或卤代c1-c24烷氧基;
所述的氧化剂为七价锰化合物。
本发明人发现,通过所述的萘酚类化合物(式1)在所述的反应溶剂体系、以及所述的氧化剂下偶联,可一步制得所述的苝醌母核结构的化合物。本发明可通过调控r1、r2基团,使制得的产物具有修饰位点,通过本发明方法可在苝醌的2/5/8/11位进行初步修饰,为接入更多官能团修饰苝醌提供了途径;例如,利用不同的供、吸电子基团来调控苝的荧光发射区域和强度。本发明方法工艺简单,初步修饰的苝醌母核结构化合物可有效解决苝醌染料的溶解性问题和无选择性修饰位点问题,为后续的二次修饰、改造提供了可能。
本发明人通过研究发现,采用所述的七价锰化合物作为氧化剂,可明显提高产物的纯度和收率;且反应条件温和、后处理简单、环境友好。
作为优选,所述的氧化剂高锰酸或高锰酸盐。
所述的氧化剂优选为高锰酸、或者为高锰酸根的水溶性盐。所述的水溶性盐例如为高锰酸根的碱金属盐、铵盐等;例如,所述的氧化剂可为高锰酸钠、高锰酸钾、高锰酸铵等。
进一步优选,所述的氧化剂为高锰酸钾。
作为优选,所述的氧化剂的投加摩尔量为式1化合物的0.05-0.5倍。
进一步优选,所述的氧化剂的投加摩尔量为式1化合物的0.20-0.28倍。
本发明人发现,氧化剂的投加当量对制得的产物的纯度以及收率均有一定的影响,通过研究发现,投加的氧化剂(以高锰酸根离子计)为式1化合物的0.20-0.28当量,最优选0.25当量时;可防止产物的过氧化;进而有助于提升产物的产率和纯度。
作为优选,所述的氧化剂以水溶液的形式投加,所述的氧化剂的水溶液中,高锰酸根的摩尔浓度为0.01~0.1mol/l。采用所述摩尔浓度的氧化剂的水溶液更利于反应温和进行,可有助于进一步防止过度氧化而破坏产物,进而进一步提高产物的纯度和收率。
进一步优选,所述的氧化剂的水溶液中,高锰酸根的摩尔浓度为0.05mol/l。
本发明人还发现,所述的反应溶剂体系对制备的产物的纯度及收率具有一定的影响。
作为优选,氧化偶联过程中,体系中的有机溶剂和水的体积比为1∶1~2。
本发明中,氧化偶联体系中的水包含外加的水以及氧化剂水溶液中引入的水;控制氧化偶联体系中的有机溶剂和水的体积比在1∶1~2之间,可提升产物的收率。
本发明人发现,调控氧化偶联反应体系的氧化剂起始溶摩尔浓度对产物的收率具有一定的影响。
作为优选,控制氧化剂的起始摩尔浓度为1.5~20mmol/l;进一步优选为3.5~8mmol/l。
本发明中,氧化偶联反应体系为有机溶剂的水溶液。
作为优选,所述的有机溶剂为可与水混溶的溶剂。
作为优选,所述的有机溶剂为四氢呋喃、c1~4醇的至少一种。
作为优选,所述的c1~4醇为碳数为1~4的单元醇、多元醇;更进一步优选为甲醇、乙醇。
作为优选,所述的有机溶剂为四氢呋喃、甲醇、乙醇中的至少一种;进一步优选为四氢呋喃。
更进一步优选,氧化偶联过程在四氢呋喃-水的混合液中进行,其中,四氢呋喃-水的体积比为1∶1~2。
作为优选,所述的r1、r2选自相同基团。
选用相同基团的物料可进一步降低产物纯化难度。
作为优选,所述的r1、r2为h或br。
本发明人研究还发现,根据式1原料调整反应溶剂体系的thf/水的比例,以及氧化剂的起始摩尔浓度;可进一步提升产物的纯度和收率。
作为优选,r1、r2为h,氧化偶联体系中,所述的有机溶剂的水溶液中,有机溶剂与水的体积比为1∶2;氧化剂的起始摩尔浓度为3.15~5.45mmol/l;所述的氧化剂的投加摩尔量为式1化合物的0.20-0.28倍。
作为优选,r1、r2为br,所述的四氢呋喃的水溶液中,四氢呋喃与水的体积比为1∶1;氧化剂的起始摩尔浓度为3.5~8mmol/l;所述的氧化剂的投加摩尔量为式1化合物的0.20-0.28倍。
选取不同的原料,在优选的溶剂体系下反应,可有助于提升产物的收率及纯度。
氧化偶联的反应温度优选在室温下进行。
本发明中,在所述的优选的反应溶剂体系及氧化剂的投加量的协同下,优选的反应时间为24~72h;进一步优选为48h。
本发明中,对氧化的反应液进行酸析处理,使目的产物沉淀;随后经过固液分离得到目的产物粗品。
酸析过程所选用的酸例如可为无机无氧化性强酸水溶液,例如盐酸。
本发明中,氧化反应后,将稀盐酸投加至反应液中,使体系的ph降至酸性;随后静置析出固体;再进行过滤处理,并采用乙酸乙酯和/或乙醚溶剂对过滤的固体进行洗涤;制得目的产物粗品。
作为优选,所述的稀盐酸中,h+的摩尔浓度为1~3mol/l。
采用酸液调整反应液的ph至低于或等于7;进一步优选为6~7;并在所述的ph下析出目的产物沉淀。
本发明人通过研究发现,摸索出一种重结晶纯化方法,无需通过色谱纯化,操作简单,生产效率高,且产品的纯度及收率高。
作为优选,采用的结晶溶剂为乙醇和/或甲醇;进一步优选为乙醇。
所述的乙醇优选为无水乙醇。
结晶过程中,结晶溶剂与目的产物粗品的液/固体积重量60-85ml/g。
结晶过程中,采用所述的结晶溶剂将所述的目的产物粗品溶剂,随后在60℃以下析晶。
本发明中,将所述的重结晶的产物固液分离后,再经干燥处理,得到所述的苝醌染料。
作为优选,将过滤得到的重结晶的产物在45~55℃下真空干燥。
本发明的制备方法工艺过程简单、合成效率高、成本低廉,利于工业化生产。此外,本发明利用醇水体系作为反应溶剂,符合环境友好规则。四溴苝醌具有2,5,8,11四个活性位点,能够轻易在该活性位点加以修饰,大大开拓了苝醌类染料的发展与应用。
本发明提供了一种化学合成方法,工艺简单,目的产物的收率可高达92%。另外,本发明可通过原料的选择,使制得的产物具有可修饰的位点,为后续苝母核化合物的修改、改造提供了一种很好的途径。
附图说明
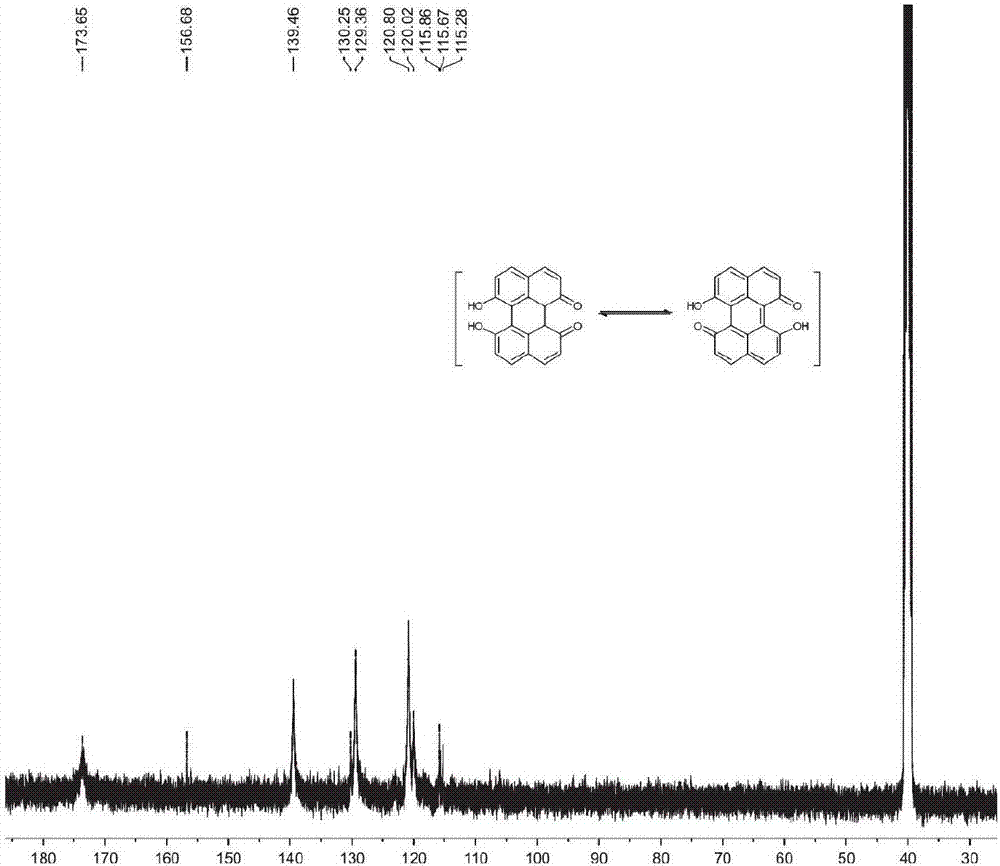
图1是苝醌的核磁共振氢谱。
图2是苝醌的核磁共振碳谱。
图3是四溴苝醌的核磁共振氢谱。
图4是四溴苝醌的核磁共振碳谱。
图5是苝醌的紫外可见光谱图。
图6是四溴苝醌的紫外可见光谱图。
图7是苝醌的红外光谱谱图。
图8是四溴苝醌的红外光谱谱图。
具体实施方式
以下实施例中,除特别申明外,所述的室温是5-15℃。
实施例1
苝醌的合成
向盛有2,7-二羟基萘(0.50g,3.12mmol)中加入50mlthf,搅拌至其完全溶解,再加入84.4ml水,向其中滴加0.05mol/l的高锰酸钾溶液15.6ml(0.25eqv),在室温下反应2天得到反应混合物。向反应混合物中加入50ml2mol/l稀盐酸,调节ph至ph=6-7。静置、过滤,分别用乙酸乙酯和乙醚各冲洗滤饼三次,得到粗产品457mg,再用30ml无水乙醇重结晶,过滤,在真空干燥箱中50℃烘干,得到435mg紫黑色固体(苝醌)。反应产率为88.7%。制得的产品的光谱测试结果参考各相关附图,其中,图1是苝醌的核磁共振氢谱。图2是苝醌的核磁共振碳谱。图5是苝醌的紫外可见光谱图。图7是苝醌的红外光谱谱图。
采用核磁共振和紫外可见法证明紫黑色物质是苝醌。1hnmr(400mhz,dmso-d6)d(ppm):7.92-7.93(d,j=6.4hz,4h),6.90-6.91(d,j=5.2hz,4h);13cnmr(100mhz,dmso-d6)d(ppm):δ173.65,156.68,139.46,130.25,129.36,120.80,120.02,115.86,115.67,115.28。
对比例1
和实施例1相比,区别在于,采用仅添加thf,不额外添加所述水:具体为:
向盛有2,7-二羟基萘(0.50g,3.12mmol)中加入134.4ml四氢呋喃,搅拌至其完全溶解,向其中滴加0.05mol/l的高锰酸钾溶液15.6ml(0.25eqv),在室温下反应2天得到反应混合物。向反应混合物中加入50ml2mol/l稀盐酸,调节ph至ph=6-7。静置、过滤,分别用乙酸乙酯和乙醚各冲洗滤饼三次,得到粗产品31mg,再用2ml无水乙醇重结晶,过滤,在真空干燥箱中50℃烘干,得到27mg紫黑色固体。反应产率为5.5%。
对比例2
和实施例1相比,区别在于,thf与水的体积比例为1∶3,具体为:
向盛有2,7-二羟基萘(0.50g,3.12mmol)中加入,37ml四氢呋喃,搅拌至其完全溶解,再加入95.4ml水,向其中滴加0.05mol/l的高锰酸钾溶液15.6ml(0.25eqv),在室温下反应2天得到反应混合物。向反应混合物中加入50ml2mol/l稀盐酸,调节ph至ph=6-7。静置、过滤,分别用乙酸乙酯和乙醚各冲洗滤饼三次,得到粗产品59mg,再用4ml无水乙醇重结晶,过滤,在真空干燥箱中50℃烘干,得到50mg紫黑色固体。反应产率为10.0%。
实施例2
四溴苝醌的合成
向盛有3,6-二溴-2,7-二羟基萘(0.50g,1.57mmol)中加入50mlthf,搅拌至其完全溶解,再加入42.1ml水,向其中滴加0.05mol/l的高锰酸钾溶液7.9ml(0.25eqv),在室温下反应2天得到反应混合物。向反应混合物中加入30ml2mol/l稀盐酸,调节ph至溶液ph=6-7。静置、过滤,分别用乙酸乙酯和乙醚各冲洗滤饼三次,得到粗产品482mg,再用35ml无水乙醇重结晶,过滤,得到460mg四溴苝醌。反应产率为92.9%。制得的产品的光谱测试结果参考各相关附图,其中,图3是四溴苝醌的核磁共振氢谱。图4是四溴苝醌的核磁共振碳谱。图6是四溴苝醌的紫外可见光谱图。图8是四溴苝醌的红外光谱谱图。
采用核磁共振和紫外可见法证明蓝黑色物质是四溴苝醌。1hnmr(400mhz,dmso-d6)d(ppm):8.32(s,2h),8.23(s,2h);13cnmr(100mhz,dmso-d6)d(ppm):179.57,163.49,144.51,137.40,136.51,128.01,125.58,117.09,116.17,113.20。
实施例3
和实施例2相比,区别仅在于,高锰酸钾溶液的浓度为0.1mol/l(0.5eqv);其他参数与实施例2相同。反应产率为45.5%。
实施例4
和实施例2相比,区别仅在于,高锰酸钾溶液的浓度为0.01mol/l(0.05eqv);其他参数与实施例2相同。反应产率为56.3%。
对比例3
和实施例2相比,区别仅在于,高锰酸钾溶液的浓度为0.5mol/l(2.5eqv);其他参数与实施例2相同。反应产率为16.8%。
对比例4
和实施例2相比,区别仅在于,高锰酸钾溶液的浓度为0.005mol/l(0.025eqv);其他参数与实施例2相同。反应产率为27.5%。
实施例5
四溴苝醌的合成
向盛有3,6-二溴-2,7-二羟基萘(0.50g,1.57mmol)中加入50ml甲醇,搅拌至其完全溶解,再加入42.1ml水,向其中滴加0.05mol/l的高锰酸钾溶液7.9ml(0.25eqv),在室温下反应2天得到反应混合物。向反应混合物中加入30ml2mol/l稀盐酸,调节ph至溶液ph=6-7。静置、过滤,分别用乙酸乙酯和乙醚各冲洗滤饼三次,得到粗产品466mg,再用35ml无水乙醇重结晶,过滤,得到433mg四溴苝醌。反应产率为87.4%。
通过各实施例及对比例发现,在本发明所述的反应体系、氧化剂的投加当量下,目标产物的收率高。例如,反应体系中的有机溶剂和水的体积比在1∶1~2下,目的产物收率较高,不在该比例范围下的目的产物收率下降较明显。研究还发现,氧化剂的投加当量对目的产物的收率具有较大影响,超出0.5当量或者少于0.05当量,目的产物的收率均不理想。此外,控制反应体系中氧化剂的起始浓度在合适的范围内,有助于进一步提升产物的收率。
- 还没有人留言评论。精彩留言会获得点赞!