一种稀土离子双掺杂Bi4P2O11发光材料及其制备方法与流程
本发明属于稀土离子双掺杂双根离子制备发光材料
技术领域:
,具体为一种稀土离子双掺杂bi4p2o11发光材料及其制备方法。
背景技术:
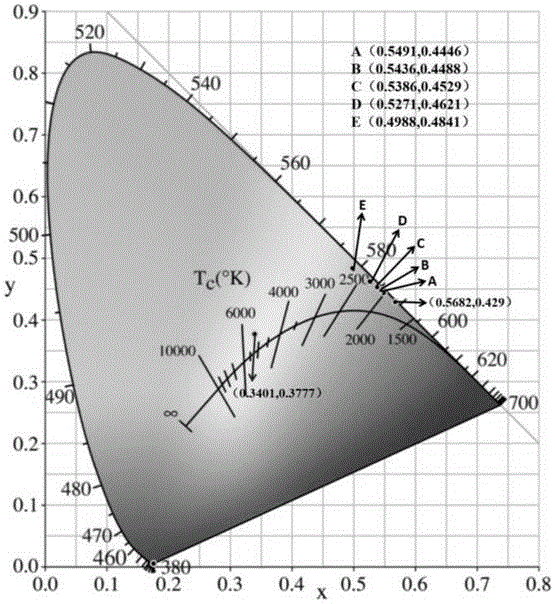
:稀土材料的发光是由于稀土离子的4f电子在f-f组态之内或f-d组态之间的跃迁而产生的。稀土发光材料由于具有发光谱带窄、色纯度高、光吸收能力强、转换效率高、物理和化学性质稳定、低毒性等特点已经被广泛应用在照明显示、信息存储放大、医学诊断和生物荧光探针等领域。双稀土离子双掺杂于某种适当的基质材料中会产生较好的能量传递效应,这样不仅能提高发光材料的荧光性能,还能达到光色可调的效果,从而进一步拓展该类发光材料的应用领域与范围。由于bi4p2o11的晶体结构与具有良好物理化学稳定性的一类磷酸盐相近,bi3+半径与稀土离子半径相近,因此bi4p2o11可作为一种重要的荧光粉基质材料。采用稀土离子特征性发光的sm3+和发光亮度、余辉时间均较优的dy3+双掺杂于基质材料bi4p2o11中,利用稀土离子间的能量传递效应和稀土离子与bi4p2o11间的协同作用,使bi4p2o11在掺杂后发光性能进一步提高,但目前关于sm3+和dy3+双掺杂于bi4p2o11基质材料还未见报道。技术实现要素:针对现有技术中存在的问题,本发明提供一种稀土离子双掺杂bi4p2o11发光材料及其制备方法,操作方便,合成工艺简单,产物粒度均匀,且合成粒度可控,无污染,成本低,使bi4p2o11在掺杂后发光性能进一步提高并实现光色可调,拓展了bi4p2o11在掺杂后的应用领域。本发明是通过以下技术方案来实现:一种稀土离子双掺杂bi4p2o11发光材料的制备方法,包括以下步骤,步骤1,将bi2o3、nh4h2po4、sm2o3和dy2o3均匀混合,研磨后得到稀土离子双掺杂bi4p2o11发光材料的前驱体原料;其中,bi2o3、sm2o3和dy2o3摩尔数的总和与nh4h2po4的摩尔数相同,sm2o3占bi2o3、sm2o3和dy2o3摩尔数总和的摩尔百分数为0.5%~10%,dy2o3占bi2o3、sm2o3和dy2o3摩尔数总和的摩尔百分数为1%~8%;步骤2,将步骤1得到的前驱体原料在600℃~1000℃下反应4h~10h后降至室温,得到一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:x%sm3+/y%dy3+,其中x为0.5~10,y为1~8。优选的,还包括步骤3,将步骤2得到的发光材料bi4p2o11:x%sm3+/y%dy3+进行研磨。优选的,还包括步骤3,将步骤2得到的发光材料bi4p2o11:x%sm3+/y%dy3+研磨0.5h~2h得到均匀的粉末。优选的,步骤1中sm2o3占bi2o3、sm2o3和dy2o3摩尔数总和的摩尔百分数为0.5%、1%、2%、3%、5%、7%或10%。优选的,sm2o3占bi2o3、sm2o3和dy2o3摩尔数总和的摩尔百分数为2%,dy2o3占bi2o3、sm2o3和dy2o3摩尔数总和的摩尔百分数为1%、2%、3%、4%、5%、6%、7%或8%。优选的,步骤2中的温度以5℃/min的速率上升至反应温度,反应后以3℃/min的降温速率降至60℃后,自然冷却至室温。优选的,步骤2中的前驱体原料在900℃下反应4h后降至室温。一种由上述任意一项所述的方法制备得到的稀土离子双掺杂bi4p2o11发光材料bi4p2o11:x%sm3+/y%dy3+。与现有技术相比,本发明具有以下有益的技术效果:本发明采用物理化学性能均稳定的bi4p2o11晶体作为稀土离子掺杂的基质材料,利用高温固相法,在高温条件下sm2o3、dy2o3、bi2o3与nh4h2po4反应得到了一种稀土离子双掺杂bi4p2o11的荧光粉bi4p2o11:x%sm3+/y%dy3+,不仅利用了sm3+与dy3+间的能量传递效应,还利用了sm3+/dy3与基质材料bi4p2o11间的协同作用,通过稀土离子sm3+/dy3间的能量转移机制和以bi4p2o11基质为主的晶体场环境的共同作用,从而大大提高了sm3+/dy3+双掺杂bi4p2o11后材料的发光性能,并实现光色可调功能。附图说明图1为对比例1所制备的荧光粉bi4p2o11:2%sm3+的xrd图。图2为sm3+浓度为0.5%、1%、2%、3%、5%、7%和10%下单掺杂所制备的荧光粉bi4p2o11:x%sm3+于599nm处测得的激发光谱图,其中x分别为0.5、1、2、3、5、7和10。图3为sm3+浓度为0.5%、1%、2%、3%、5%、7%和10%下单掺杂所制备的荧光粉bi4p2o11:x%sm3+于激发波长为403nm处测得的发射光谱图,其中x分别为0.5、1、2、3、5、7和10。图4为对比例2所制备的荧光粉bi4p2o11:5%dy3+于激发波长为350nm处测得的发射光谱图。图5为实施例2所制备的荧光粉bi4p2o11:2%sm3+/4%dy3+的xrd图。图6为实施例1-3、实施例10和实施例13所制备的dy3+浓度为1%、2%、4%、6%和8%下的荧光粉bi4p2o11:2%sm3+/x%dy3+在599nm监测波长处的激发光谱图,其中x分别为1、2、4、6和8。图7为实施例1-3、实施例10和实施例13所制备的dy3+浓度为1%、2%、4%、6%和8%下的荧光粉bi4p2o11:2%sm3+/x%dy3+在激发波长为403nm监测波长处的发射光谱图,其中x分别为1、2、4、6和8。图8为实施例1-3、实施例10和实施例13所制备的dy3+浓度为1%、2%、4%、6%和8%下的荧光粉bi4p2o11:2%sm3+/x%dy3+在574nm监测波长下的激发光谱图,其中x分别为1、2、4、6和8。图9为实施例1-3、实施例10和实施例13所制备的dy3+浓度为1%、2%、4%、6%和8%下的荧光粉bi4p2o11:2%sm3+/x%dy3+在激发波长为388nm监测波长下的发射光谱图,其中x分别为1、2、4、6和8。图10为对比例1-2、实施例1-3、实施例10和实施例13所制备的荧光粉样品的cie色度坐标图。具体实施方式下面结合具体的实施例对本发明做进一步的详细说明,所述是对本发明的解释而不是限定。本发明提供一种稀土离子双掺杂bi4p2o11发光材料及其制备方法,其中的制备方法具体包括以下步骤,步骤1,按照化学计量比分别称取一定量的sm2o3、dy2o3、bi2o3与nh4h2po4,其中的bi2o3、sm2o3和dy2o3摩尔数的总和与nh4h2po4的摩尔数相同,sm2o3占bi2o3、sm2o3和dy2o3摩尔数总和的摩尔百分数为0.5%~10%,dy2o3占bi2o3、sm2o3和dy2o3摩尔数总和的摩尔百分数为1%~8%,将称取好的bi2o3、nh4h2po4、sm2o3和dy2o3混合均匀,之后在玛瑙研钵中进行研磨,将研磨好的前驱体原料放进氧化铝陶瓷坩埚中;步骤2,将盛有前驱体原料的氧化铝陶瓷坩埚放进高温箱式电阻炉中,在600℃~1000℃高温条件下对其进行固相反应,焙烧温度以5℃/min的速率上升,反应时间设置为4h~10h,待反应完成后降温,降温速率为3℃/min,降至60℃后自然冷却至室温,然后将装有产物样品的坩埚从电阻炉中取出;步骤3,将取出的样品产物用玛瑙研钵充分研磨0.5h~2h成细致均匀的粉末后即得一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:x%sm3+/y%dy3+,其中x为0.5~10,y为1~8。在固相反应中,粉末的粒度大多在微米量级,反应物通过颗粒接触面在晶格中扩散、成核和生长;在该反应过程中,反应前驱物sm2o3、dy2o3、bi2o3与nh4h2po4以粉末形态均匀混合,它们之间以分子或离子接触,依靠高温条件下键的断裂、键的形成和原子间的重新排列生成新的物质bi4p2o11:x%sm3+/y%dy3+;该反应不仅利用了sm3+与dy3+间的能量传递效应,还利用了sm3+/dy3与基质材料bi4p2o11间的协同作用,通过稀土离子sm3+/dy3间的能量转移机制和以bi4p2o11基质为主的晶体场环境的共同作用,从而大大提高了sm3+/dy3+双掺杂bi4p2o11后材料的发光性能,并实现光色可调功能。一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:x%sm3+/y%dy3+利用上述制备方法制得。下面通过具体的实施例对本发明做进一步详细说明。实施例1一种稀土离子双掺杂bi4p2o11发光材料的制备方法,具体包括以下步骤,步骤1,将1.81g的bi2o3、0.46g的nh4h2po4、0.028g的sm2o3和0.015g的dy2o3进行均匀混合得到前驱体原料,此时bi2o3的摩尔数为3.88mmol,nh4h2po4的摩尔数为4mmol,sm2o3的摩尔数为0.08mmol,dy2o3的摩尔数为0.04mmol,因此sm3+的掺杂浓度为2%,dy3+的掺杂浓度为1%,之后在玛瑙研钵中进行研磨,将研磨好的原料放进氧化铝陶瓷坩埚中;步骤2,将盛有前驱体原料的氧化铝陶瓷坩埚放进高温箱式电阻炉中,设置焙烧温度以5℃/min的速率上升,在600℃高温条件下固相反应10h,降温速率为3℃/min,降至60℃后自然冷却至室温,然后将装有产物样品的坩埚从电阻炉中取出;步骤3,将取出的样品产物用玛瑙研钵充分研磨0.5h成细致均匀的粉末后即得一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:2%sm3+/1%dy3+。一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:2%sm3+/1%dy3+利用上述制备方法制得。实施例2一种稀土离子双掺杂bi4p2o11发光材料的制备方法,具体包括以下步骤,步骤1,将1.75g的bi2o3、0.46g的nh4h2po4、0.028g的sm2o3和0.06g的dy2o3进行均匀混合得到前驱体原料,此时bi2o3的摩尔数为3.76mmol,nh4h2po4的摩尔数为4mmol,sm2o3的摩尔数为0.08mmol,dy2o3的摩尔数为0.16mmol,因此sm3+的掺杂浓度为2%,dy3+的掺杂浓度为4%,之后在玛瑙研钵中进行研磨,将研磨好的原料放进氧化铝陶瓷坩埚中;步骤2,将盛有前驱体原料的氧化铝陶瓷坩埚放进高温箱式电阻炉中,设置焙烧温度以5℃/min的速率上升,在900℃高温条件下固相反应4h,降温速率为3℃/min,降至60℃后自然冷却至室温,然后将装有产物样品的坩埚从电阻炉中取出;步骤3,将取出的样品产物用玛瑙研钵充分研磨2h成细致均匀的粉末后即得一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:2%sm3+/4%dy3+。一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:2%sm3+/4%dy3+利用上述制备方法制得。实施例3一种稀土离子双掺杂bi4p2o11发光材料的制备方法,具体包括以下步骤,步骤1,将1.68g的bi2o3、0.46g的nh4h2po4、0.028g的sm2o3和0.12g的dy2o3进行均匀混合得到前驱体原料,此时bi2o3的摩尔数为3.6mmol,nh4h2po4的摩尔数为4mmol,sm2o3的摩尔数为0.08mmol,dy2o3的摩尔数为0.32mmol,因此sm3+的掺杂浓度为2%,dy3+的掺杂浓度为8%,之后在玛瑙研钵中进行研磨,将研磨好的原料放进氧化铝陶瓷坩埚中;步骤2,将盛有前驱体原材料的氧化铝陶瓷坩埚放进高温箱式电阻炉中,设置焙烧温度以5℃/min的速率上升,在1000℃高温条件下固相反应7h,降温速率为3℃/min,降至60℃后自然冷却至室温,然后将装有产物样品的坩埚从电阻炉中取出;步骤3,将取出的样品产物用玛瑙研钵充分研磨1h成细致均匀的粉末后即得一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:2%sm3+/8%dy3+。一种稀土离子双掺杂bi4p2o11发光材料bi4p2o11:2%sm3+/8%dy3+利用上述制备方法制得。实施例4-14本发明中不同实施例的差别在于sm3+的掺杂浓度、dy3+的掺杂浓度、反应温度、反应时间和研磨时间不同,因此将实施例4-14中与实施例1-3不同的参数列成表格,具体如表1所示,其中sm2o3、dy2o3和bi2o3的单位均为g,sm3+和dy3+的掺杂浓度单位均为%,反应温度的单位为℃,反应时间和研磨时间的单位均为h。表1实施例4-14中的具体参数实施例sm2o3dy2o3bi2o3sm3+dy3+反应温度反应时间研磨时间40.0070.0601.780.546804.3150.0140.1051.71177008.5160.0420.0751.71357506270.0700.0151.755195050.580.0940.1201.62789804.5190.1400.0451.621039305.52100.0280.0301.792264090.5110.0280.0451.77236609.51.5120.0280.0751.732572080.5130.0280.0901.71268007.51.5140.0280.1051.69278506.51.5为了充分说明sm3+和dy3+双掺杂bi4p2o11发光材料的特性,现将其与sm3+和dy3+分别单掺杂bi4p2o11发光材料做对比,对比例1-2如下:对比例1sm3+单掺杂bi4p2o11发光材料的制备方法包括以下步骤,步骤1,将1.83g的bi2o3、0.46g的nh4h2po4和0.028g的sm2o3进行均匀混合得到前驱体原料,此时bi2o3的摩尔数为3.92mmol,nh4h2po4的摩尔数为4mmol,sm2o3的摩尔数为0.08mmol,因此sm3+的掺杂浓度为2%,之后在玛瑙研钵中进行研磨,将研磨好的原料放进氧化铝陶瓷坩埚中;步骤2,将盛有前驱体原材料的氧化铝陶瓷坩埚放进高温箱式电阻炉中,设置焙烧温度以5℃/min的速率上升,在900℃高温条件下固相反应4h,降温速率为3℃/min,降至60℃后自然冷却至室温,然后将装有产物样品的坩埚从电阻炉中取出;步骤3,将取出的样品产物用玛瑙研钵充分研磨1h成细致均匀的粉末后即得sm3+单掺杂bi4p2o11发光材料bi4p2o11:2%sm3+。图1是bi4p2o11:2%sm3+荧光粉的xrd图和标准卡对比谱图。通过对比分析看出制备所得bi4p2o11:2%sm3+荧光粉样品的衍射峰与标准卡片jcpds46-0420的衍射峰相吻合,没有杂峰出现,表明所制备样品为纯bi4p2o11晶体,且由图能看出少量sm3+的掺杂并没有引起bi4p2o11:2%sm3+晶型的改变。因此认为当bi3+被sm3+取代后不会引起样品晶型发生改变。图2是浓度为0.5%、1%、2%、3%、5%、7%和10%下sm3+单掺杂所制备的bi4p2o11:x%sm3+样品在监测波长为599nm处测得的激发光谱图。在346nm、363nm、376nm、403nm、418nm和441nm处均有吸收峰,分别对应6h5/2→4h9/2、6h5/2→4d3/2、6h5/2→4p7/2、6h5/2→4k11/2、6h5/2→(4p,6p)5/2和6h5/2→4g9/2的跃迁,其中sm3+的最佳激发波长为403nm,对应sm3+基态的6h5/2向高能级的4k11/2跃迁。图3是激发波长为403nm处测得的浓度为0.5%、1%、2%、3%、5%、7%和10%下sm3+单掺杂所制备的bi4p2o11:x%sm3+的发射光谱图,可以看出存在四个发射峰,表明sm3+能够吸收波长为403nm的光子跃迁到高能级进而辐射跃迁发射出波长为560nm、599nm、646nm和720nm的光子,其中599nm处的发射峰强度最大,560nm、599nm、646nm和720nm分别对应sm3+的4g5/2→6h5/2,4g5/2→6h7/2,4g5/2→6h9/2和4g5/2→6h11/2跃迁,发射峰强度随掺杂浓度先升高后降低,最强发射峰为2%的sm3+掺杂浓度,过量的掺杂会导致浓度淬灭现象发生。对比例2dy3+单掺杂bi4p2o11发光材料的制备方法包括以下步骤,步骤1,将1.77g的bi2o3、0.46g的nh4h2po4和0.075g的dy2o3进行均匀混合得到前驱体原料,此时bi2o3的摩尔数为3.8mmol,nh4h2po4的摩尔数为4mmol,dy2o3的摩尔数为0.2mmol,因此dy3+的掺杂浓度为5%,之后在玛瑙研钵中进行研磨,将研磨好的原料放进氧化铝陶瓷坩埚中;步骤2,将盛有前驱体原材料的氧化铝陶瓷坩埚放进高温箱式电阻炉中,设置焙烧温度以5℃/min的速率上升,在900℃高温条件下固相反应4h,降温速率为3℃/min,降至60℃后自然冷却至室温,然后将装有产物样品的坩埚从电阻炉中取出;步骤3,将取出的样品产物用玛瑙研钵充分研磨2h成细致均匀的粉末后即得dy3+单掺杂bi4p2o11发光材料bi4p2o11:5%dy3+。图4是所得bi4p2o11:5%dy3+样品在激发光波长为350nm处测得的发射光谱图,由图看出有480nm和574nm的发射峰,分别对应4f9/2→6h15/2和4f9/2→6h13/2跃迁,最强发射峰位于480nm。图5为实施例2制备的bi4p2o11:2%sm3+/4%dy3+荧光粉样品的xrd图谱,可以看出样品的衍射峰仍然与jcpds46-0420标准卡片一致,说明双掺杂sm3+和dy3+稀土离子所制备的样品与图1中单掺杂sm3+的样品和无掺杂纯相bi4p2o11衍射峰保持一致,双掺杂sm3+和dy3+的产物没有影响其物相和晶体结构。图6是实施例1-3、实施例10和实施例13所得dy3+浓度为1%、2%、4%、6%和8%下的荧光粉bi4p2o11:2%sm3+/x%dy3+样品在599nm监测波长下测得的激发光谱图。从图中可以看出双掺杂的样品与图2单掺杂bi4p2o11:x%sm3+样品在599nm监测波长下所测得的激发光谱的峰位和峰型均没有发生变化,位于346nm、363nm、376nm、403nm、418nm和441nm处的激发峰分别对应6h5/2→4h9/2、6h5/2→4d3/2、6h5/2→4p7/2、6h5/2→4k11/2、6h5/2→(4p,4p)5/2和6h5/2→4g9/2的跃迁,最强激发峰在403nm。图7是实施例1-3、实施例10和实施例13所得dy3+浓度为1%、2%、4%、6%和8%下的荧光粉bi4p2o11:2%sm3+/x%dy3+样品在激发波长为403nm处测得的发射光谱图。从图中可以看出双掺杂的样品与图3单掺杂bi(4-x)smxp2o11样品在403nm激发波长下测得的发射峰的仍没有发生变化,位于560nm、599nm、646nm和720nm分别对应sm3+的4g5/2→6h5/2,4g5/2→6h7/2,4g5/2→6h9/2和4g5/2→6h11/2跃迁。固定sm3+的掺杂浓度为2%时,掺杂浓度为1%的dy3+掺杂样品的发射峰强最大,随着dy3+掺杂浓度增加到2%,4%,6%和8%时发射峰强呈下降趋势。图8是实施例1-3、实施例10和实施例13所得dy3+掺杂浓度为1%、2%、4%、6%和8%下的bi4p2o11:2%sm3+/x%dy3+样品在574nm监测波长下的激发光谱图。从图中可以看出位于353nm、365nm、388nm、430nm、455nm和474nm分别对应6h15/2→4k15/2、6h15/2→6p7/2、6h15/2→6p5/2、6h15/2→4i13/2、6h15/2→4g11/2和6h15/2→4i15/2跃迁,最强激发峰位于388nm。图9是实施例1-3、实施例10和实施例13所得dy3+掺杂浓度为1%、2%、4%、6%和8%下制备的bi4p2o11:2%sm3+/x%dy3+样品在388nm监测波长下测得的的发射光谱图。将双掺杂的样品与图4单掺杂bi4p2o11:5%dy3+样品的发射峰进行对比发现,位于480nm和574nm处的发射峰峰位和峰型没有发生变化,其对应4f9/2→6h15/2和4f9/2→6h13/2的跃迁,最强发射峰为574nm;然而发现在599nm处多出了一个发射峰,由图4可知599nm处的发射峰本来源于sm3+单掺杂于bi4p2o11基质中的发射峰,说明在双掺杂的bi4p2o11:2%sm3+/x%dy3+样品中存在sm3+向dy3+的能量传递现象。另外,固定sm3+掺杂浓度为2%时,发射峰强度随dy3+掺杂浓度先增加后减小,分析过量的掺杂导致了dy3+浓度淬灭。图10是对比例1-2、实施例1-3、实施例10和实施例13所制备荧光粉样品的cie色度,表2为制备荧光粉样品的cie色度坐标。如图10和表2所示,对于对比例2单掺杂dy3+制备的bi4p2o11:5%dy3+样品来讲,cie色度坐标(0.3401,0.3777)落在接近白光区域,而对于对比例1单掺杂sm3+制备的bi4p2o11:2%sm3+样品来讲,cie色度坐标(0.5682,0.429)处于红色区域,当将dy3+和sm3+两种离子进行双掺杂制备的bi4p2o11:2%sm3+/x%dy3+样品,其中x分别为1、2、4、6和8,对这些样品来讲,随着dy3+掺杂浓度的改变,其色坐标会由橙红光逐渐变成黄光,具有光色可调功能。具体地,当dy3+掺杂浓度为1%时,样品的色度坐标落在橙红光区域,即图10中a处;当dy3+掺杂浓度提高到2%时,其色度坐标逐渐向黄光区域偏移,即图10中b处;当dy3+掺杂浓度持续增大到4%时,其色度坐标向黄光区域进一步偏移,即图10中c处;当dy3+掺杂浓度增大到6%时,其色度坐标落在图10中d处;当dy3+掺杂浓度增大到8%时,其色度坐标已经位于黄光区域,即图10中e处;图中样品色度坐标的逐渐偏移,表明采用制备的样品具有光色可调功能。表2对比例1-2、实施例1-3、实施例10和实施例13所制备荧光粉样品的cie色度坐标本发明采用固相反应法,利用sm2o3、dy2o3、bi2o3与nh4h2po4反应合成发光材料bi4p2o11:2%sm3+/x%dy3+,按照一定的摩尔百分比进行反应,反应完成后待自然冷却至室温,研磨即得该样品粉体。本发明采用简单易行的固相合成法,制备一种稀土离子钐/镝双掺杂bi4p2o11发光材料,利用稀土离子之间的能量转换和稀土离子与bi4p2o11基质间的协同作用,可获得一种具有光色可调功能的高光效bi4p2o11:2%sm3+/x%dy3+发光材料,可广泛应用于多种领域需求。以上所述仅为本发明的若干种实施方式,不是全部的实施方式,本领域普通技术人员通过阅读上述实施例而对本发明技术方案采取的任何等效的变换均为本发明具体实施方式所涵盖的实施方式。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种稀土掺杂陶瓷基金刚石基板材料及其制备方法
- 通过稀土化合物共掺杂可控析出ML-Ag团簇和Ag纳米晶的发光玻璃及其制备方法
- 丝网印刷用稀土上转换发光油墨及其制备方法及防伪应用
- 提高稀土离子掺杂无机氟化物上转换发光强度的方法
- 一种稀土离子掺杂的铌酸锆荧光粉及其制备方法
- 一种稀土离子掺杂的红色荧光粉及其制备方法
- 多壁碳纳米管/ Ce<sup>3+</sup>掺杂TiO<sub>2</sub>纳米纤维在对苯酚催化降解中的应用
- 一种Er<sup>3+</sup>/Nd<sup>3+</sup>共掺杂Na<sub>5</sub>Lu<sub>9</sub>F<sub>32</sub>单晶体及其生长方法
- 一种具有红光定向发射性能的镱/铥离子共掺六方相氟钇化钠微米晶体的制作方法
- 一种具有红光定向发射性能的β-NaYF4