链中部具有官能团的二烯弹性体和包含其的橡胶组合物的制作方法
由于节省燃料和环保需要已经成为重点,希望制备滞后性尽可能低的混合物从而能够将其加工成用于制造参与组成外胎的各种半成品(例如底层、胎侧或胎面)的橡胶组合物的形式并且获得具有降低滚动阻力的轮胎。
理想地,例如轮胎胎面必须满足大量技术要求,这些技术要求本质上通常相互矛盾,包括增加的耐磨损性同时为轮胎提供低滚动阻力。
此外,还必须实现混合物滞后性的降低(混合物滞后性的降低标志着滚动阻力的降低)同时保持混合物特别是在原料态下的适加工性,而且还要维持弹性体的耐流动性。
已经研究了许多解决方案从而实现降低滞后性的目标。可以特别提及的是,在聚合结束时通过官能化剂、偶联剂或星形支化剂改变二烯聚合物和共聚物的结构,目的是获得由此改性的聚合物和填料(无论是炭黑还是增强无机填料)之间的良好相互作用。
对于包含无机增强填料(例如二氧化硅)的混合物,提出使用通过烷氧基硅烷衍生物官能化的二烯聚合物,特别是通过组合或不组合通过烷氧基硅烷官能化进行的官能化和通过其它官能团(特别是胺、亚胺、环氧化物或硫醇官能团)进行的官能化。
因此,文献WO 2007047943 A1描述了通过使活性弹性体与带有保护性硫醇官能团的烷氧基硅烷化合物反应从而在聚合物链的端部进行官能化,目的是改进包含官能化弹性体的经增强橡胶组合物的滞后性。
在文献EP 0 692 492 A1和EP 0 692 493 A1中,申请人描述了通过带有环氧化物官能团的烷氧基硅烷化合物在二烯弹性体链的端部和中部进行官能化,目的是改进包含官能化弹性体的经增强橡胶组合物的增强和滞后性。
专利文献中广泛记载通过带有氨基官能团的烷氧基硅烷化合物进行官能化。在旨在制造轮胎的橡胶组合物中,链端部通过带有氨基的烷氧基硅烷官能团官能化的弹性体能够与二氧化硅和炭黑(或甚至是这两种填料的混合物)同样良好地组合。
申请人在文献WO 2009133068 A1中描述了一种基本上由弹性体的偶联实体组成的官能化的二烯弹性体,所述弹性体的链中具有带有烷氧基硅烷官能团和胺官能团的基团,该基团的硅原子键合二烯弹性体链的两个部分。这种在链中部被官能化的弹性体赋予包含其的组合物改进的机械性质和动态性质,特别是改进的滞后性,同时保持令人满意的原料态加工性,目的是特别用于轮胎胎面。
除了改进滞后性(造成轮胎滚动阻力的降低)之外,抗磨性(能够评估轮胎的耐磨性)的增加是旨在用于制造轮胎的橡胶组合物的另一个希望性能。
为了改进轮胎的耐磨性和抗磨性,已知希望胎面中具有一定刚度。然而,经验表明这种胎面刚度经常(适当情况下通常过多地)不利地影响滚动阻力性质,因为其伴随着橡胶组合物的滞后性损失的明显增加。
此外,用于制造轮胎的材料的设计者持续关注于改进橡胶组合物的机械性质和动态性质的折衷,目的是改进包含所述橡胶组合物的轮胎的性能而不会不利地影响其制备阶段或弹性体性质。因此,例如,性质折衷的改进不能破坏弹性体的流动,否则会造成橡胶的运输和储存过程中的主要问题。
过去曾经提出加入使用包含锡或硅的额外的偶联剂或星形支化剂偶联或星形支化的弹性体从而限制流动。然而,过去提出的组合不能始终另经增强的橡胶组合物的加工性/滞后性折衷满足轮胎应用。
本发明旨在解决的技术问题是针对轮胎应用进一步改进橡胶组合物的滞后性和抗磨性的折衷,而不会不利地影响其加工或该组合物中包含的弹性体的性质,特别是其抗流动性。
本发明人在其研发过程中出人意料地发现通过经改性的二烯弹性体实现了该目的,所述经改性的二烯弹性体包含:相对于经改性的二烯弹性体的总重量至少70重量%的线型二烯弹性体,所述线型二烯弹性体主要在链中部被任选部分或完全水解从而形成硅烷醇的烷氧基硅烷基团官能化,所述烷氧基硅烷基团任选带有其它官能团(适当的情况下优选为伯胺、仲胺或叔胺官能团),所述烷氧基硅烷通过硅原子键合至二烯弹性体;和相对于经改性的二烯弹性体的总重量至多30重量%的星形支化的二烯弹性体,所述经改性的二烯弹性体的门尼粘度为50至80,并且其玻璃化转变温度(Tg)为-100℃至-80℃,赋予包含所述经改性的二烯弹性体的橡胶组合物改进的原料态加工性/滞后性/磨损折衷同时保持弹性体的抗流动性。
本发明的主题因此是经改性的二烯弹性体,其包含:
a)相对于经改性的二烯弹性体的总重量至少70重量%的线型二烯弹性体,所述线型二烯弹性体主要在链中部被任选部分或完全水解从而形成硅烷醇的烷氧基硅烷基团官能化,所述烷氧基硅烷基团任选带有其它官能团并且硅原子位于二烯弹性体的主链中,和
b)相对于经改性的二烯弹性体的总重量大于0且至多30重量%的星形支化的二烯弹性体,
所述经改性的二烯弹性体的门尼粘度为50至80,并且玻璃化转变温度为-100℃至-80℃,优选-95℃至-80℃。
本发明的另一个主题是经增强的橡胶组合物,其基于至少一种增强填料和弹性体基质,所述弹性体基质包含至少一种所述经改性的二烯弹性体。
在本说明书中,除非另有声明,显示的所有百分比(%)为重量%。此外,由表述“在a和b之间”表示的任何数值范围代表由大于a延伸至小于b的数值范围(即不包括极限a和b),而由表述“a至b”表示的任何数值范围意指由a延伸直至b的数值范围(即包括严格极限a和b)。
表述“组合物基于”应被理解为意指组合物包含所用的各种成分的混合物和/或反应产物,在制备组合物的各个阶段的过程中,特别是在其交联或硫化的过程中,这些基本成分中的一些能够反应或旨在至少部分地彼此反应。
在本申请中,与化合物相关的术语“主要地”或“主要的”被理解为意指在组合物的相同类型的化合物中,该化合物是主要的化合物,即在相同类型的化合物中,其是占据最大重量份的化合物。因此,在构成二烯弹性体的官能实体中,以官能化二烯弹性体的总重量计,官能化的二烯弹性体的“主要的”官能实体占据最大重量份。在仅包含一种特定类型的化合物的体系中,所述化合物属于本发明意义上的主要的化合物。
在本说明书中,Tg旨在表示化合物(特别是本发明的经改性的二烯弹性体)根据标准ASTM D3418测得的玻璃化转变温度。
在本说明书中,门尼粘度旨在表示化合物(特别是本发明的经改性的二烯弹性体)根据标准ASTM D1646测得的ML(1+4)100℃门尼粘度。
在本说明书中,伯胺或仲胺旨在表示通过本领域技术人员已知的保护性基团保护或未保护的伯胺或仲胺。
在本说明书中,术语“经改性的二烯弹性体”应理解为意指包含具有一个或多个杂原子的基团的二烯弹性体。
该基团可以位于线型弹性体主链的端部。则可以说二烯弹性体在链端被官能化。其通常为通过活性弹性体与官能化剂的反应获得的弹性体,亦即任何至少单官能的分子,官能团为本领域技术人员已知的与活性链端反应的任何类型的化学基团。
该基团可以位于线型弹性体主链中。则可以说二烯弹性体在链中部(不同于“链端”的位置)偶联或官能化,尽管基团不精确位于弹性体链的中部。其通常为通过活性弹性体的两条链与官能化剂的反应获得的弹性体,亦即任何至少双官能的分子,官能团为本领域技术人员已知的与活性链端反应的任何类型的化学基团。
该基团可以位于中心,其上键合n个弹性体链(n>2),形成星形支化结构。则可以说二烯弹性体为星形支化的。其通常为通过活性弹性体的n条链与星形支化剂的反应获得的弹性体,亦即任何多官能的分子,官能团为本领域技术人员已知的与活性链端反应的任何类型的化学基团。
本领域技术人员将理解,与包含多于一个相对于活性弹性体具有反应性的官能团的试剂的官能化反应造成链端和在链中部被官能化的实体(构成官能化弹性体的线型链)以及任选的星形支化的实体的混合物。根据操作条件(主要是官能化剂与活性链的摩尔比),某些实体在混合物中是主要的。
术语“二烯弹性体”应当以已知的方式被理解为意指至少部分(即均聚物或共聚物)得自二烯单体(带有两个共轭或非共轭碳-碳双键的单体)的(一种或多种)弹性体。更具体地,二烯弹性体旨在表示通过具有4至12个碳原子的共轭二烯单体的聚合获得的任何均聚物,或者通过一种或多种共轭二烯相互间或与一种或多种具有8至20个碳原子的乙烯基芳族化合物共聚而获得的任何共聚物。
根据一些变体形式,相对于弹性体重量,二烯弹性体包含95重量%至100重量%的二烯单元和0至5重量%的乙烯基芳族单元。
在共聚物的情况下,相对于共聚物的总重量,其包含95重量%至小于100重量%,优选96重量%至99重量%的二烯单元和大于0至5重量%,优选1重量%至4重量%的乙烯基芳族单元。
如下特别适合作为根据本发明的方法中可以使用的共轭二烯:1,3-丁二烯、2-甲基-1,3-丁二烯、2,3-二(C1-C5-烷基)-1,3-丁二烯(例如2,3-二甲基-1,3-丁二烯、2,3-二乙基-1,3-丁二烯、2-甲基-3-乙基-1,3-丁二烯或2-甲基-3-异丙基-1,3-丁二烯)、苯基-1,3-丁二烯、1,3-戊二烯和2,4-己二烯等。
如下特别适合作为乙烯基芳族化合物:苯乙烯、邻-、间-或对-甲基苯乙烯、“乙烯基甲苯”商用混合物、对-(叔丁基)苯乙烯、甲氧基苯乙烯、乙烯基均三甲基苯、二乙烯基苯和乙烯基萘等。
二烯弹性体优选选自高度不饱和的二烯弹性体,包括聚丁二烯(BR)、合成聚异戊二烯(IR)、丁二烯共聚物,特别是丁二烯和乙烯基芳族单体的共聚物,异戊二烯共聚物和这些弹性体的混合物。这种共聚物更特别是丁二烯/苯乙烯共聚物(SBR)、异戊二烯/丁二烯共聚物(BIR)、异戊二烯/苯乙烯共聚物(SIR)和异戊二烯/丁二烯/苯乙烯共聚物(SBIR)。在这些聚合物中,丁二烯均聚物和丁二烯/苯乙烯共聚物(SBR)是特别优选的。
特别适合作为丁二烯均聚物的是聚丁二烯,特别是1,2-单元的含量(摩尔%)在4%和20%之间(优选10%至15%)并且顺式-1,4-/反式-1,4-摩尔比为1至0.65的那些。
特别适合作为丁二烯/苯乙烯共聚物的是相对于共聚物重量的苯乙烯含量在0和5重量%之间(更特别是1重量%至4重量%)并且丁二烯部分的1,2-键含量(摩尔%)在4%和20%之间(优选10%至15%)的丁二烯/苯乙烯共聚物。
二烯弹性体可以具有取决于所使用的聚合条件的任何微观结构。该共聚物可以为嵌段、无规、序列或微序列弹性体等,并且可以在分散体或溶液中制备。在基于二烯和芳族乙烯基的共聚物(特别是包含丁二烯和苯乙烯)的情况下,两种单体优选无规分布。
这些弹性体的微结构由极性剂的存在或不存在以及阴离子聚合步骤中使用的极性剂的量决定。优选地,当二烯弹性体基于二烯和苯乙烯时,在聚合步骤中使用一定量的极性剂从而促进苯乙烯沿着聚合物链的无规分布同时将1,2-键的含量保持在4%和20%之间,优选10%至15%。
在链中部被官能化的二烯弹性体a)和星形支化的二烯弹性体b)可以在官能化和星形支化之前具有相同的微观结构或不同的微观结构。
优选地,主要在链中部被官能化的线型二烯弹性体a)和星形支化的二烯弹性体b)在官能化和星形支化之前具有相同的微观结构。
还优选地,主要在链中部被官能化的线型二烯弹性体a)和星形支化的二烯弹性体b)在官能化和星形支化之前具有相同的微观结构和相同的宏观结构。
根据本发明,术语“主要在链中部被官能化的线型二烯弹性体”(即弹性体a))旨在表示官能化弹性体的线型实体(即在链端被官能化的线型链和在链中部被官能化的线型链)的混合物,在链中部被官能化的线型链在该混合物中占据主要重量,并且优选占弹性体a)的总重量的至少50重量%,还更优选至少80重量%。
根据一个优选的实施方案,根据本发明的经改性的二烯弹性体包含相对于经改性的二烯弹性体的总重量至少75重量%或甚至至少80重量%的主要在链中部被官能化的线型二烯弹性体a)。
根据另一个优选的实施方案,根据本发明的经改性的二烯弹性体包含相对于经改性的二烯弹性体的总重量至多25重量%或甚至至多20重量%的星形支化的二烯弹性体b)。
根据另一个优选的实施方案,根据本发明的经改性的二烯弹性体包含相对于经改性的二烯弹性体的总重量至少5重量%的星形支化的二烯弹性体b)。
根据改进了原料态加工性/滞后性/磨损折衷同时保持了弹性体的抗流动性的特别优选的实施方案,根据本发明的经改性的二烯弹性体包含相对于经改性的二烯弹性体的总重量至少75重量%的主要在链中部被官能化的线型二烯弹性体a)和相对于经改性的二烯弹性体的总重量至多25重量%的星形支化的二烯弹性体b)。更特别地,根据本发明的经改性的二烯弹性体包含相对于经改性的二烯弹性体的总重量至少5重量%的星形支化的二烯弹性体b)。
星形支化的二烯弹性体b)优选为基于锡或基于硅的星形支化的二烯弹性体。
星形支化的二烯弹性体b)优选为包含三个或四个支链的星形支化的二烯弹性体。
根据本发明的一个特别优选的实施方案,星形支化的二烯弹性体b)为通过包含硅原子的基团支化的具有三个支链的星形支化的弹性体,所述硅原子带有或不带有能够与增强填料相互作用的其它官能团,所述硅原子被二烯弹性体的三个支链取代。硅原子上的取代基有利地与二烯弹性体a)的取代基相同,因为这样可以使用单种官能化剂。
根据本发明的第一个变体形式,除了包含直接键合至弹性体链的硅原子并且任选部分或完全水解形成硅烷醇的烷氧基硅烷基团之外,二烯弹性体a)不包含其它官能团。
根据本发明的第二个变体形式,二烯弹性体a)还包含能够与增强填料相互作用的其它官能团,该官能团有利地被烷氧基硅烷基团的硅直接带有或经由间隔基团带有。这被理解为不排除如下事实:包含直接键合至弹性体链的硅原子的烷氧基硅烷基团有利地与增强填料相互作用。
术语“有利地与增强填料相互作用的烷氧基硅烷基团”或“能够与增强填料相互作用的官能团”被理解为意指在由填料增强的橡胶组合物内能够与所述填料形成物理键或化学键的任何其它烷氧基硅烷基团或官能团。在所述官能团和填料上存在的官能团之间可以例如通过共价键、氢键、离子键和/或静电键建立该相互作用。
烷氧基硅烷基团的烷氧基可以具有式R’O-,其中R’表示未取代或经取代的C1-C10(甚至是C1-C8)烷基,优选C1-C4烷基,更优选甲基和乙基。
术语“能够与增强填料相互作用的官能团”优选旨在表示包含至少一个选自N、S、O或P的杂原子的官能团。在这些官能团中,例如可以提及环状或非环状伯胺、仲胺或叔胺、异氰酸酯、亚胺、氰基、硫醇、羧酸酯、环氧化物或伯膦、仲膦或叔膦。
作为仲胺或叔胺官能团,因此可以提及被C1-C10(优选C1-C4)烷基(更优选甲基或乙基)取代的胺,或形成包含氮原子和至少一个碳原子(优选2至6个碳原子)的杂环的环状胺。例如合适的是甲基氨基、二甲基氨基、乙基氨基、二乙基氨基、丙基氨基、二丙基氨基、丁基氨基、二丁基氨基、戊基氨基、二戊基氨基、己基氨基、二己基氨基或六亚甲基氨基基团,优选二乙基氨基和二甲基氨基基团。
作为亚胺官能团,可以提及酮亚胺。例如,合适的是(1,3-二甲基亚丁基)氨基、(亚乙基)氨基、(1-甲基亚丙基)氨基、(4-N,N-二甲基氨基亚苄基)氨基、(亚环己基)氨基、二氢咪唑基团和咪唑基团。
作为羧酸酯官能团,因此可以提及丙烯酸酯或甲基丙烯酸酯。所述官能团优选为甲基丙烯酸酯。
作为环氧化物官能团,可以提及环氧基团或环氧丙氧基团。
作为仲膦或叔膦官能团,可以提及被C1-C10(优选C1-C4)烷基(更优选甲基或乙基)取代的膦,或二苯基膦。例如,合适的是甲基膦基、二甲基膦基、乙基膦基、二乙基膦基、乙基甲基膦基和二苯基膦基。
根据本发明的第二个变体形式,能够与增强填料相互作用的其它官能团可以直接键合至硅原子,所述硅原子本身直接键合至二烯弹性体。
根据本发明的第二个变体形式,能够与增强填料相互作用的其它官能团和直接键合至二烯弹性体的硅原子可以通过间隔基团连接在一起,所述间隔基团可以是原子或原子基团。间隔基团可以是饱和或不饱和的、环状或非环状的、线型或支化的二价C1-C18脂族烃类基团或二价C6-C18芳族烃类基团,并且可以包含一个或多个芳族基团和/或一个或多个杂原子。烃类基团可以任选被取代。
优选地,间隔基团为线型或支化的二价C1-C18脂族烃类基团,更优选线型二价烃类基团,还更优选线型二价C2或C3烃类基团。
特别和能够与增强填料相互作用的其它官能团的性质、间隔基团的性质、二烯弹性体的性质、包含硅原子的官能团的性质和不同实体的比例相关的上述不同的优选或非优选方面可以彼此组合,只要它们彼此相容。
非常优选地,根据二烯弹性体a)还包含能够与增强填料相互作用的其它官能团的第二个变体形式,能够与增强填料相互作用的其它官能团为伯胺、仲胺或叔胺。由于改进了滞后性质,使用本发明的该第二个变体形式是特别有利的。
优选地,能够与增强填料相互作用的官能团为叔胺官能团,更优选为二乙胺或二甲胺。
根据本发明的有利的变体形式,满足如下特征中的至少任一个、至少两个、至少三个、至少四个、至少五个和优选全部:
-星形支化的二烯弹性体b)为通过包含硅原子(所述硅原子带有能够与增强填料相互作用的其它官能团)的基团支化的具有三个支链的星形支化的弹性体,
-能够与增强填料相互作用的其它官能团为叔胺,更特别是二乙基氨基或二甲基氨基基团,
-间隔基团为线型C1-C18烃类基团,还更优选为线型C2或C3烃类基团,
-烷氧基硅烷基团为任选部分或完全水解从而形成硅烷醇的甲氧基硅烷或乙氧基硅烷,
-二烯弹性体为丁二烯聚合物,更特别是丁二烯均聚物或丁二烯/苯乙烯共聚物,
-相对于经改性的二烯弹性体的总重量,经改性的二烯弹性体包含至少75重量%的主要在链中部被官能化的线型二烯弹性体a)和至多25重量%的星形支化的二烯弹性体b)。
因此非常优选地,根据本发明的经改性的弹性体为二烯弹性体,其中:
-星形支化的二烯弹性体b)为通过包含硅原子(所述硅原子带有二乙基氨基或二甲基氨基基团)的基团支化的具有三个支链的星形支化的弹性体,
-间隔基团为线型C3烃类基团,
-烷氧基硅烷基团为任选部分或完全水解从而形成硅烷醇的甲氧基硅烷或乙氧基硅烷,
-二烯弹性体为丁二烯均聚物或丁二烯/苯乙烯共聚物,
-相对于经改性的二烯弹性体的总重量,经改性的二烯弹性体包含至少75重量%的主要在链中部被官能化的线型二烯弹性体a)和至多25重量%的星形支化的二烯弹性体b)。
根据本发明的经改性的二烯弹性体可以通过下述方法获得。
根据本发明的任一个变体形式,经改性的二烯弹性体的该制备方法的第一步骤是至少一种共轭二烯单体在聚合引发剂的存在下进行阴离子聚合。
作为聚合引发剂,可以使用任何已知的单官能阴离子引发剂。但是,优选使用包括碱土金属,例如锂的引发剂。
包含碳-锂键的有机锂引发剂是特别合适的。优选使用不包含杂原子的烃类有机锂引发剂。代表性的化合物为脂族有机锂化合物,例如乙基锂、正丁基锂(n-BuLi)、异丁基锂等。包含胺-锂键的有机锂引发剂也是合适的。代表性的化合物为由环状仲胺获得的氨基锂,例如吡咯烷和六亚甲基亚胺。
聚合优选在例如为脂族或脂环族烃(例如戊烷、己烷、庚烷、异辛烷、环己烷或甲基环己烷)或者芳族烃(例如苯、甲苯或二甲苯)的惰性烃类溶剂的存在下进行。
聚合可以连续或分批进行。
聚合通常在20℃和150℃之间,优选在30℃至120℃附近的温度下进行。当然,也可能在聚合结束时加入金属转移剂,用于改变活性链端部的反应性。
得自聚合的活性二烯弹性体然后官能化从而制备根据本发明的经改性的二烯弹性体。
根据本发明的经改性的二烯弹性体的制备的第一个变体形式,主要在链中部被官能化的线型二烯弹性体a)和星形支化的二烯弹性体b)以合适的比例混合。
在链中部被官能化的二烯弹性体a)可以有利地通过活性链端与能够在链中部引入烷氧基硅烷基团(所述烷氧基硅烷基团任选部分或完全水解从而形成硅烷醇并且带有或不带有其官能团)的偶联剂的反应获得;在适当的情况下,当其它官能团为胺时,可以特别根据专利申请WO2009133068A1中描述的步骤实现二烯弹性体a)的链中部的官能化,该文献的描述以引用的方式并入本文。
星形支化的二烯弹性体b)可以以本身已知的方式通过活性链端与星形支化剂的反应获得,亦即任何多官能的分子,官能团为本领域技术人员已知的与活性链端反应的任何类型的化学基团。
两种弹性体的混合可以在例如为脂族或脂环族烃(例如戊烷、己烷、庚烷、异辛烷或环己烷)或者芳族烃(例如苯、甲苯或二甲苯)的惰性溶剂中进行,所述惰性溶剂可以与聚合溶剂相同。混合则优选在20℃和120℃之间,优选在30℃至110℃附近的温度下进行。
根据本发明的经改性的二烯弹性体的制备的第二个变体形式,源自聚合步骤的活性二烯弹性体经受星形支化剂的反应和偶联剂的反应,所述偶联剂能够通过用二烯弹性体的一个或两个链取代硅原子从而在聚合物链的中部引入烷氧基硅烷基团,所述烷氧基硅烷基团可以水解形成硅烷醇或不水解并且任选带有能够与橡胶组合物内的增强填料相互作用的其它官能团。
根据本发明的这两个合成变体形式可以使用的偶联剂带有烷氧基硅烷官能团(所述烷氧基硅烷官能团可水解从而形成硅烷醇官能团或者是不可水解的烷氧基硅烷官能团)和能够与增强填料相互作用的任选其它官能团,两个官能团直接彼此键合或者经由间隔基团键合。能够与增强填料相互作用的官能团和间隔基团如上定义。
偶联剂可以通过下式(I)表示:
其中:
-Y为饱和或不饱和的、环状或非环状的二价C1-C18脂族烃类基团或C6-C18芳族烃类基团,优选线型或支化的二价C1-C18脂族烃类基团,更优选线型二价脂族烃类基团,还更优选线型C2或C3烃类基团,
-X为氢原子或能够与增强填料相互作用的官能团,
-R’基团被取代或未被取代并且相同或不同,表示C1-C10甚至是C1-C8烷基,优选C1-C4烷基,更优选甲基和乙基。
能够与增强填料相互作用的官能团如上定义。
能够与增强填料相互作用的官能团优选为伯胺、仲胺或叔胺官能团。
在伯胺的情况下,氮原子则可以被两个保护性基团取代,所述保护性基团特别是两个三烷基甲硅烷基,烷基具有1至4个碳原子。
在仲胺的情况下,氮原子则可以被一个保护性基团取代,所述保护性基团特别是三烷基甲硅烷基(其中烷基具有1至4个碳原子)和C1-C10优选C1-C4烷基,更优选甲基或乙基。
在叔胺的情况下,氮原子则可以被两个保护性基团取代,所述两个保护性基团相同或不同,可能是C1-C10优选C1-C4烷基,更优选甲基或乙基,或者氮的两个取代基与所述氮形成包含氮原子和至少一个碳原子(优选2至6个碳原子)的杂环。
作为偶联剂,可以例如提及烷基三烷氧基硅烷、(N,N-二烷基氨基丙基)三烷氧基硅烷、(N-烷基氨基丙基)三烷氧基硅烷(其仲胺官能团受三烷基甲硅烷基的保护)和(氨基丙基)三烷氧基硅烷(其伯胺官能团受两个三烷基甲硅烷基的保护)。
优选地,偶联剂可以选自(3-N,N-二甲基氨基丙基)三甲氧基硅烷、(3-N,N-二甲基氨基丙基)三乙氧基硅烷、(3-N,N-二乙基氨基丙基)三甲氧基硅烷、(3-N,N-二乙基氨基丙基)三乙氧基硅烷、(3-N,N-二丙基氨基丙基)三甲氧基硅烷、(3-N,N-二丙基氨基丙基)三乙氧基硅烷、(3-N,N-二丁基氨基丙基)三甲氧基硅烷、(3-N,N-二丁基氨基丙基)三乙氧基硅烷、(3-N,N-二戊基氨基丙基)三甲氧基硅烷、(3-N,N-二戊基氨基丙基)三乙氧基硅烷、(3-N,N-二己基氨基丙基)三甲氧基硅烷、(3-N,N-二己基氨基丙基)三乙氧基硅烷、(3-六亚甲基氨基丙基)三甲氧基硅烷、(3-六亚甲基氨基丙基)三乙氧基硅烷、(3-吗啉代丙基)三甲氧基硅烷、(3-吗啉代丙基)三乙氧基硅烷、(3-哌啶基丙基)三甲氧基硅烷或(3-哌啶基丙基)三乙氧基硅烷。更优选地,偶联剂为(3-N,N-二甲基氨基丙基)三甲氧基硅烷。
优选地,偶联剂可以选自(3-N,N-甲基三甲基甲硅烷基氨基丙基)三甲氧基硅烷、(3-N,N-甲基三甲基甲硅烷基氨基丙基)三乙氧基硅烷、(3-N,N-乙基三甲基甲硅烷基氨基丙基)三甲氧基硅烷、(3-N,N-乙基三甲基甲硅烷基氨基丙基)三乙氧基硅烷、(3-N,N-丙基三甲基甲硅烷基氨基丙基)三甲氧基硅烷或(3-N,N-丙基三甲基甲硅烷基氨基丙基)三乙氧基硅烷。更优选地,偶联剂为(3-N,N-甲基三甲基甲硅烷基氨基丙基)三甲氧基硅烷。
优选地,偶联剂可以选自(3-N,N-双(三甲基甲硅烷基)氨基丙基)三甲氧基硅烷和(3-N,N-双(三甲基甲硅烷基)氨基丙基)三乙氧基硅烷。更优选地,偶联剂为(3-N,N-双(三甲基甲硅烷基)氨基丙基)三甲氧基硅烷。
有利地,当能够与增强填料相互作用的官能团为胺官能团时,胺官能团为叔胺官能团并且偶联剂则优选为(3-N,N-二甲基氨基丙基)三甲氧基硅烷。
能够与增强填料相互作用的官能团还可以是异氰酸酯官能团。优选地,官能化剂则选自(3-异氰酸根合丙基)三甲氧基硅烷和(3-异氰酸根合丙基)三乙氧基硅烷。
能够与增强填料相互作用的官能团还可以是亚胺官能团。优选地,官能化剂则选自N-(1,3-二甲基亚丁基)-3-(三甲氧基甲硅烷基)-1-丙胺、N-(1,3-二甲基亚丁基)-3-(三乙氧基甲硅烷基)-1-丙胺、N-(1,3-甲基亚乙基)-3-(三甲氧基甲硅烷基)-1-丙胺、N-(1,3-甲基亚乙基)-3-(三乙氧基甲硅烷基)-1-丙胺、N-亚乙基-3-(三甲氧基甲硅烷基)-1-丙胺、N-亚乙基-3-(三乙氧基甲硅烷基)-1-丙胺、N-(1-甲基亚丙基)-3-(三甲氧基甲硅烷基)-1-丙胺、N-(1-甲基亚丙基)-3-(三乙氧基甲硅烷基)-1-丙胺、N-(4-N,N-二甲基氨基亚苄基)-3-(三甲氧基甲硅烷基)-1-丙胺、N-(4-N,N-二甲基氨基亚苄基)-3-(三乙氧基甲硅烷基)-1-丙胺、N-(亚环己基)-3-(三甲氧基甲硅烷基)-1-丙胺、N-(亚环己基)-3-(三乙氧基甲硅烷基)-1-丙胺、N-(3-三甲氧基甲硅烷基丙基)-4,5-二氢咪唑、N-(3-三乙氧基甲硅烷基丙基)-4,5-二氢咪唑、N-(3-三甲氧基甲硅烷基丙基)-4,5-咪唑或N-(3-三乙氧基甲硅烷基丙基)-4,5-咪唑。
能够与增强填料相互作用的官能团还可以是氰基官能团。优选地,官能化剂则选自(3-氰基丙基)三甲氧基硅烷和(3-氰基丙基)三乙氧基硅烷。
能够与增强填料相互作用的官能团还可以是受保护或未受保护的硫醇官能团。例如可以提及(S-三烷基甲硅烷基巯基烷基)三烷氧基硅烷。优选地,官能化剂则选自(S-三甲基甲硅烷基巯基丙基)三甲氧基硅烷、(S-三甲基甲硅烷基巯基丙基)三乙氧基硅烷、(S-叔丁基二甲基甲硅烷基巯基丙基)三甲氧基硅烷、(S-叔丁基二甲基甲硅烷基巯基丙基)三乙氧基硅烷、(S-三甲基甲硅烷基巯基乙基)三甲氧基硅烷、(S-三甲基甲硅烷基巯基乙基)三乙氧基硅烷、(S-叔丁基二甲基甲硅烷基巯基乙基)三甲氧基硅烷或(S-叔丁基二甲基甲硅烷基巯基乙基)三乙氧基硅烷。
能够与增强填料相互作用的官能团还可以是羧酸酯官能团。作为羧酸酯官能团,可以提及丙烯酸酯或甲基丙烯酸酯。所述官能团优选为甲基丙烯酸酯。优选地,官能化剂则选自(3-甲基丙烯酰氧基丙基)三甲氧基硅烷和(3-甲基丙烯酰氧基丙基)三乙氧基硅烷。
能够与增强填料相互作用的官能团还可以是环氧化物官能团。优选地,官能化剂则选自(2-环氧丙氧基乙基)三甲氧基硅烷、(2-环氧丙氧基乙基)三乙氧基硅烷、(3-环氧丙氧基丙基)三甲氧基硅烷、(3-环氧丙氧基丙基)三乙氧基硅烷、2-(3,4-环氧基环己基)乙基三甲氧基硅烷或2-(3,4-环氧基环己基)乙基三乙氧基硅烷。
能够与增强填料相互作用的官能团还可以是受保护或未受保护的伯膦官能团,受保护或未受保护的仲膦官能团或叔膦官能团。优选地,官能化剂则选自(3-P,P-双三甲基甲硅烷基膦基丙基)三甲氧基硅烷、(3-P,P-双三甲基甲硅烷基膦基丙基)三乙氧基硅烷、(3-甲基三甲基甲硅烷基膦基丙基)三甲氧基硅烷、(3-甲基三甲基甲硅烷基膦基丙基)三乙氧基硅烷、(3-乙基三甲基甲硅烷基膦基丙基)三甲氧基硅烷、(3-乙基三甲基甲硅烷基膦基丙基)三乙氧基硅烷、(3-二甲基膦基丙基)三甲氧基硅烷、(3-二甲基膦基丙基)三乙氧基硅烷、(3-二乙基膦基丙基)三甲氧基硅烷、(3-二乙基膦基丙基)三乙氧基硅烷、(3-乙基甲基膦基丙基)三甲氧基硅烷、(3-乙基甲基膦基丙基)三乙氧基硅烷、(3-二苯基膦基丙基)三甲氧基硅烷或(3-二苯基膦基丙基)三乙氧基硅烷。
偶联剂与活性聚合物链的引发剂的摩尔比为0.30至0.80,优选0.40至0.65,还更优选0.45至0.55。
根据本发明的经改性的二烯弹性体的制备的任一个变体形式,星形支化剂优选为官能度大于2的基于锡或基于硅的试剂。所述星形支化剂是本领域技术人员已知的。它们不仅包括三卤化锡和硅化合物例如SnR1X’3、SnHX’3、SiR1X’3、SiHX’3,而且包括SnX’4、SiX’4或甚至是烷氧基化的硅化合物,特别是被能够与增强填料相互作用的官能团被取代或未被取代的三烷氧基化硅烷。在这些式中,R1为具有1至20个碳原子的烷基或芳烷基并且X’为卤素优选Cl,并且能够与增强填料相互作用的官能团如上定义。
根据本发明的经改性的二烯弹性体的制备的第二个变体形式的特别有利的实施方案,偶联剂和星形支化剂是同一种化合物。根据该有利的实施方案,官能化可以连续进行并且特别根据申请WO 2015018599 A1中描述的步骤进行。
根据本发明的经改性的二烯弹性体的制备的第二个变体形式的另一个实施方案,偶联剂和星形支化剂不同。根据该具体实施方案,得自聚合步骤的活性二烯弹性体的官能化可以在20℃至120℃的温度下,首先在合适量的星形支化剂的存在下进行,从而星形支化至多30重量%的活性二烯弹性体。然后,通过加入能够在聚合物链的中部引入烷氧基硅烷基团的偶联剂并且与该试剂反应使得二烯弹性体的剩余的活性链官能化,所述烷氧基硅烷基团带有或不带有能够与橡胶组合物内的增强填料相互作用的官能团,优选伯胺、仲胺或叔胺。
根据本发明的官能化剂带有受保护的伯胺官能团或受保护的仲胺官能团的变体形式,合成方法之后可以继续进行伯胺或仲胺的去保护步骤。该步骤在改性反应之后进行。通过受保护的胺基官能化的链可以例如与酸、碱、氟化衍生物(例如四丁基氟化铵)、银盐(例如硝酸银)等反应从而使该胺官能团去保护。这些不同方法描述于T.W.Green和P.G.M.Wuts的文章“Protective Groups in Organic Synthesis”(第三版,1999)。该去保护步骤的作用是使经改性的二烯弹性体的可水解烷氧基硅烷官能团全部或部分水解从而使其转化成硅烷醇官能团。
根据本发明的官能化剂带有受保护的硫醇官能团的变体形式,合成方法之后可以继续进行硫醇的去保护步骤。该步骤在改性反应之后进行。通过受保护的硫醇基团官能化的链可以例如与水、醇或酸(盐酸、硫酸、羧酸)反应。该去保护步骤的作用是使经改性的二烯弹性体的可水解烷氧基硅烷官能团全部或部分水解从而使其转化成硅烷醇官能团。
根据本发明的官能化剂带有受保护的伯膦或仲膦官能团的变体形式,合成方法之后可以继续进行膦的去保护步骤。该步骤在改性反应之后进行。通过受保护的膦基团官能化的链可以例如与水、醇或酸(盐酸、硫酸、羧酸)反应。该去保护步骤的作用是使经改性的二烯弹性体的可水解烷氧基硅烷官能团全部或部分水解从而使其转化成硅烷醇官能团。
根据本发明的变体形式,合成方法可以包括通过加入酸、碱或中性化合物使得可水解烷氧基硅烷官能团水解的步骤,如文献EP 2 266 819 A1中所述。可水解官能团则全部或部分转化成硅烷醇官能团。至少50摩尔%,甚至至少80摩尔%,且至多100摩尔%的官能团可以因此而水解。
根据本发明的经改性的二烯弹性体的合成方法之后可以以已知的方法继续进行经改性的弹性体的回收步骤。
根据该方法的变体形式,这些步骤包括汽提步骤从而以干燥形式回收源自之前步骤的弹性体。该汽提步骤的作用是使经改性的二烯弹性体的可水解烷氧基硅烷官能团全部或部分水解从而使其转化成硅烷醇官能团。有利地,至少50摩尔%的官能团可以因此而水解。
本领域技术人员将理解这些步骤可以彼此组合,只要它们相容。因此,根据本发明的经改性的二烯弹性体的合成方法可以在改性步骤之后包括解保护步骤、特定水解步骤和汽提步骤中的全部步骤或一部分步骤。
根据本发明的经改性的二烯弹性体具有令人满意的抗流动性,在该橡胶的储存和运输的过程中具有良好稳定性。
对于轮胎应用而言,根据本发明的经改性的二烯弹性体可以有利地用在通过至少一种无机填料(例如二氧化硅)增强的橡胶组合物中从而改进其原料态加工性/滞后性/抗磨性折衷。该橡胶组合物也是本发明的主题。
因此,正如上文所解释的,本发明的另一个主题是经增强的橡胶组合物,其基于至少一种增强填料和弹性体基质,所述弹性体基质包含至少一种所述经改性的二烯弹性体。
应理解橡胶组合物可以包含一种或多种根据本发明的所述经改性的二烯弹性体。
根据本发明的增强的橡胶组合物可以以经交联状态或非交联状态(换言之可交联状态)提供。
有利地,作为根据本发明的经改性的弹性体,橡胶组合物包含的二烯弹性体满足如下特征中的至少任一个、至少两个、至少三个、至少四个、至少五个和优选全部:
-星形支化的二烯弹性体b)为通过包含硅原子(所述硅原子带有能够与增强填料相互作用的其它官能团)的基团支化的具有三个支链的星形支化的弹性体,
-能够与增强填料相互作用的其它官能团为叔胺,更特别是二乙基氨基或二甲基氨基基团,
-间隔基团为线型C1-C18烃类基团,还更优选为线型C2或C3烃类基团,
-包含硅原子的官能团为任选部分或完全水解从而形成硅烷醇的甲氧基硅烷或乙氧基硅烷,
-二烯弹性体为丁二烯聚合物,更特别是丁二烯均聚物或丁二烯/苯乙烯共聚物,
-相对于经改性的二烯弹性体的总重量,经改性的二烯弹性体包含至少75重量%的主要在链中部被官能化的线型二烯弹性体a)和至多25重量%的星形支化的二烯弹性体b)。
优选地,作为根据本发明的经改性的弹性体,橡胶组合物包含的二烯弹性体满足:
-星形支化的二烯弹性体b)为通过包含硅原子(所述硅原子带有二乙基氨基或二甲基氨基)的基团支化的具有三个支链的星形支化的弹性体,
-能够与增强填料相互作用的其它官能团为叔胺,更特别是二乙基氨基或二甲基氨基基团,
-间隔基团为线型C3烃类基团,
-烷氧基硅烷基团为任选部分或完全水解从而形成硅烷醇的甲氧基硅烷或乙氧基硅烷,
-二烯弹性体为丁二烯均聚物或丁二烯/苯乙烯共聚物,
-相对于经改性的二烯弹性体的总重量,经改性的二烯弹性体包含至少75重量%的主要在链中部被官能化的线型二烯弹性体a)和至多25重量%的星形支化的二烯弹性体b)。
根据不同的变体形式,根据本发明的经改性的二烯弹性体可以在组合物中单独使用或者与至少一种其它常规二烯弹性体(无论其是否是星形支化、偶联或官能化的)共混使用。优选地,本发明中使用的该其它二烯弹性体选自由聚丁二烯(BR)、合成聚异戊二烯(IR)、天然橡胶(NR)、丁二烯共聚物、异戊二烯共聚物和这些弹性体的混合物组成的二烯弹性体组。这种共聚物更优选选自丁二烯/苯乙烯共聚物(SBR)、乙烯/丁二烯共聚物(EBR)、异戊二烯/丁二烯共聚物(BIR)、异戊二烯/苯乙烯共聚物(SIR)和异戊二烯/丁二烯/苯乙烯共聚物(SBIR)。还有可能设想与除了二烯弹性体之外的任何合成弹性体的共混物,甚至是与除了弹性体之外的任何聚合物(例如热塑性聚合物)的共混物。
当共混使用的常规弹性体为天然橡胶和/或一种或多种二烯聚合物(例如聚丁二烯、聚异戊二烯或丁二烯/苯乙烯或丁二烯/苯乙烯/异戊二烯共聚物)时,以100重量份根据本发明的经改性的二烯弹性体计,该弹性体或这些弹性体(改性或未改性)可以以1至70重量份存在。
应注意当该组合物中的不同于本发明的经改性的二烯弹性体的弹性体的比例变得更低时,根据本发明的组合物的性质的改进更大。
因此优选地,弹性体基质以重量计主要包含根据本发明的经改性的二烯弹性体。
更优选地,弹性体基质仅由根据本发明的经改性的二烯弹性体组成。
除了如上所述的至少一种弹性体基质之外,本发明的橡胶组合物包含至少一种增强填料。
可以使用已知能够增强可用于制造轮胎胎面的橡胶组合物的任何类型的增强填料,例如炭黑,增强无机填料如二氧化硅(以已知的方式与偶联剂组合),或这两类填料的混合物。
单独使用或以混合物形式使用的所有炭黑,特别是通常用于轮胎胎面的HAF、ISAF或SAF型炭黑(“轮胎级”炭黑)适合用作炭黑。在后者中,更特别地提及100、200或300系列(ASTM级)增强炭黑,例如N115、N134、N234、N326、N330、N339、N347或N375炭黑。
在本申请中,“增强无机填料”根据定义应当被理解为任何无机或矿物填料而无论其颜色及其来源(天然或合成),其能够单独增强用于制造轮胎的橡胶组合物而无需除了中间偶联剂之外的任何方法;所述填料通常以已知的方式特征在于其表面上存在羟基(-OH)。
硅质类型的矿物填料,特别是二氧化硅(SiO2),或者铝质类型的矿物填料,特别是氧化铝(Al2O3)特别适合作为增强无机填料。所使用的二氧化硅可以是本领域技术人员公知的任何增强二氧化硅,特别是BET表面积和CTAB比表面积均小于450m2/g,优选30至400m2/g,特别是在60和300m2/g之间的任何沉淀二氧化硅或热解二氧化硅。还将提及铝质类型的矿物填料,特别是氧化铝(Al2O3)或氢氧化铝(氧化铝)或增强氧化钛,例如在US 6 610 261和US 6 747 087中描述的。还适合作为增强填料的是其它性质的增强填料,特别是炭黑,前提是这些增强填料覆盖有硅质层或者在其表面上包含官能位点(特别是羟基位点),需要使用偶联剂从而建立填料和弹性体之间的键合。例如,可以例如提及用于轮胎的炭黑,如例如专利文献WO 96/37547和WO 99/28380中所述。
以何种物理状态提供增强无机填料并不重要,无论其为粉末、微珠、颗粒、珠的形式或任何其它适当的致密化形式。当然,术语“增强无机填料”也理解为是指不同增强无机填料,特别是如上所述的高度可分散硅质填料。
优选地,总增强填料(炭黑和/或如二氧化硅的其它增强填料)的含量在10和200phr之间,更优选在30和150phr之间,还更优选在70和130phr之间,最佳值以已知的方式根据特定的目标应用而不同。
根据本发明的一个变体形式,增强填料主要不是炭黑,即增强填料包含大于增强填料总重量的50重量%的一种或多种非炭黑填料,特别是增强无机填料例如二氧化硅,或甚至仅由所述填料组成。
根据该变体形式,当还存在炭黑时,炭黑可以以小于20phr,更优选小于10phr(例如在0.5和20phr之间,特别是1至10phr)的含量使用。
根据本发明的另一个变体形式,使用主要包含炭黑并且任选包含二氧化硅或其它无机填料的增强填料。
当增强填料包含需要使用偶联剂从而建立填料和弹性体之间的键合的填料时,根据本发明的橡胶组合物通常还包含能够有效提供该键合的试剂。当二氧化硅作为增强填料存在于组合物中时,可以使用有机硅烷作为偶联剂,特别是烷氧基硅烷多硫化物或巯基硅烷,或至少双官能的聚有机硅氧烷。
所述偶联剂不能与用于合成上述经改性的二烯弹性体的偶联剂混淆。
在根据本发明的组合物中,偶联剂的含量有利地小于20phr,应理解通常希望使用尽可能少的偶联剂。其含量优选在0.5和12phr之间。偶联剂的存在取决于增强无机填料的存在。本领域技术人员容易根据该填料的含量调节偶联剂的含量,相比于除了炭黑之外的增强无机填料的量,偶联剂的含量通常为约0.5重量%至15重量%。
除了偶联剂之外,根据本发明的橡胶组合物还可以包含偶联活化剂,用于覆盖填料的试剂,或更通常的加工助剂,所述加工助剂能够以已知的方式通过改进填料在橡胶基质中的分散并且降低组合物的粘度从而改进组合物在未处理态下的可加工能力,这些试剂为例如可水解硅烷如烷基烷氧基硅烷、多元醇、聚醚、伯胺、仲胺或叔胺或者羟基化的或可水解的聚有机硅氧烷。
根据本发明的橡胶组合物还可以包含增强有机填料,所述增强有机填料可以代替全部或一部分炭黑或全部或一部分上述其它增强无机填料。作为增强有机填料的示例,可以提及如申请WO-A-2006/069792、WO-A-2006/069793、WO-A-2008/003434和WO-A-2008/003435中所述的官能化的聚乙烯基有机填料。
根据本发明的橡胶组合物还可以包含通常用于旨在制造轮胎的弹性体组合物中的全部或部分常用添加剂,例如颜料、非增强填料、保护剂(如抗臭氧蜡、化学抗臭氧剂或抗氧化剂、抗疲劳剂)、增塑剂、增强树脂或増塑树脂、亚甲基受体(例如酚醛树脂)或亚甲基给体(例如HMT或H3M)(例如描述于申请WO 02/10269)、基于硫或硫给体和/或过氧化物和/或双马来酰亚胺的交联体系、硫化促进剂或硫化活化剂。
使用本领域技术人员公知的两个连续制备阶段在适当的混合器中制造组合物:在高温(高达110℃和190℃之间,优选130℃和180℃之间的最大温度)下的热机械加工或捏合的第一阶段(“非制备”阶段),然后是在较低温度(通常小于110℃,例如在40℃和100℃之间)下的机械加工的第二阶段(“制备”阶段),在所述精制阶段的过程中引入交联体系。
根据本发明的组合物的制备方法通常包括:
(i)在130℃和180℃之间的最大温度下进行热机械加工组合物的除了交联体系之外的组分(包括根据本发明的经改性的二烯弹性体和增强填料)的第一步骤,然后
(ii)在低于所述第一步骤中的所述最大温度的温度下,进行机械加工的第二步骤,在该过程中引入所述交联体系。
在进行上述步骤(i)和(ii)之前,该方法还可以包括根据如上所述的方法制备经改性的二烯弹性体的步骤。
本发明的另一主题是由橡胶制成的轮胎半成品,其包含根据本发明的可交联或经交联的橡胶组合物或由所述组合物组成。
因此获得的最终组合物然后可以例如以片材或板材的形式进行压延或者例如挤制从而形成可以用作轮胎橡胶半成品的橡胶成型件。
由于改进的滞后性/原料态加工性/磨损折衷同时维持了弹性体(所述弹性体的特征在于根据本发明的经增强的橡胶组合物)的抗流动性,应注意所述组合物可以构成任何轮胎半成品(特别是胎面)从而特别降低其滚动阻力并且改进耐磨性。
本发明的最后主题因此为包括根据本发明的半成品(特别是胎面)的轮胎。
本发明的上述及其它特征将通过阅读如下以举例说明但并非限制性方式描述的本发明的数个示例性实施例而得以更好地理解。
实施例
所用的测量和测试
尺寸排阻色谱
SEC(尺寸排阻色谱)技术能够根据大分子的尺寸通过填充多孔凝胶的柱分离溶液中的大分子。根据大分子的流体力学体积分离它们,体积最大的首先被洗脱。
虽然不是绝对的方法,SEC能够确定聚合物的分子量分布。可以从市售标样确定各个数均分子量(Mn)和重均分子量(Mw),并且可以通过“Moore”校准计算多分散指数(Ip=Mw/Mn)。
在分析之前对聚合物样品不进行特别处理。首先将样品以大约1g.l-1的浓度简单地溶解在洗脱溶剂中。接着在注入前,使溶液通过孔隙率为0.45μm的过滤器进行过滤。
所使用的装置为Waters Alliance色谱线。洗脱溶剂为四氢呋喃或四氢呋喃+1体积%的二异丙胺+1体积%的三乙胺,流速为1ml.min-1,系统温度为35℃并且分析时间为30分钟。使用商标名为Styragel HT6E的一组两根Waters柱。注入的聚合物样品溶液的体积为100μl。检测器为Waters 2410差示折光器,并且使用色谱数据的软件为Waters Empower系统。
计算的平均分子量是相对于具有如下微观结构的SBR制得的校准曲线:25重量%(相对于聚合物重量)的苯乙烯型单元,23重量%(相对于丁二烯部分)的1,2-型单元和50重量%(相对于丁二烯部分)的反式-1,4-型单元。
高分辨率尺寸排阻色谱
使用高分辨率SEC技术确定聚合物样品中存在的各种链种群的重量百分比。
在分析之前对聚合物样品不进行特别处理。首先将样品以大约1g.l-1的浓度简单地溶解在洗脱溶剂中。接着在注入前,使溶液通过孔隙率为0.45μm的过滤器进行过滤。
所使用的装置为Waters Alliance 2695色谱线。洗脱溶剂为四氢呋喃,流速为0.2ml.min-1,体系温度为35℃。使用一组三个串联的相同柱(Shodex,长度300mm,直径8mm)。所述一组柱的理论板数大于22 000。注入的聚合物样品溶液的体积为50μl。检测器为“Waters 2414”差示折光器,并且使用色谱数据的软件为Waters Empower系统。
计算的分子量是相对于具有如下微观结构的SBR制得的校准曲线:25重量%的苯乙烯型单元,23重量%的1,2-型单元和50重量%的反式-1,4-型单元。
门尼粘度
对于聚合物和橡胶组合物,根据标准ASTM D-1646测量门尼粘度ML(1+4)100℃。
使用如标准ASTM D-1646中描述的振荡稠度计。根据如下原理进行门尼塑性测量:将原料态(即在固化之前)的弹性体或组合物在加热至100℃的圆柱形室中模制。在预热1分钟之后,转子以2转/分钟在测试试样内旋转,在旋转4分钟之后测量用于维持该运动的工作扭矩。门尼塑性ML(1+4)以“门尼单位”(MU,1MU=0.83N.m)表示。
组合物的门尼粘度和弹性体的门尼粘度之间的差异使得能够测量原料态可加工性或加工性。该差异越小,原料态加工性越好。
差示量热法
通过差示量热计(差示扫描量热计)根据标准ASTM E1356-08(2014)确定弹性体的玻璃化转变温度(Tg)。
近红外(NIR)光谱
通过近红外(NIR)光谱技术表征弹性体的微观结构。
近红外光谱(NIR)用于定量地确定弹性体中苯乙烯的重量含量及其微观结构(1,2-、反式-1,4-和顺式-1,4-丁二烯单元的相对分布)。该方法的原理基于针对多组分体系概括的Beer-Lambert定律。由于该方法是间接的,其使用通过13C NMR确定的含有组合物的标准弹性体进行多元校准[Vilmin,F.,Dussap,C.和Coste,N.,Applied Spectroscopy,2006,60,619-29]。然后从厚度大约730μm的弹性体膜的NIR光谱计算苯乙烯含量和微观结构。使用装备有由Peltier效果冷却的InGaAs检测器的Bruker Tensor 37Fourier变换近红外光谱仪,在4000和6200cm-1之间采用2cm-1的分辨率以传输模式采集光谱。
固有粘度
根据如下原理,通过弹性体在甲苯中的0.1g.dl-1溶液确定弹性体在25℃下的固有粘度:
通过测量聚合物溶液的流动时间t和毛细管中甲苯的流动时间to确定固有粘度。
在置于热静力学控制于25±0.1℃的浴中的Ubbelohde管(毛细管直径为0.46mm,容量为18至22ml)中测量甲苯的流动时间和0.1g.dl-1的聚合物溶液的流动时间。
根据如下关系式获得固有粘度:
其中:
C:聚合物的甲苯溶液的浓度(用g.dl-1表示),
t:聚合物的甲苯溶液的流动时间(用秒表示),
to:甲苯的流动时间(用秒表示),
ηinh:固有粘度(用dl.g-1表示)。
冷流(CF(1+6)100℃)
其涉及到测量在固定条件(100℃)下在给定时间(6小时)内通过校准模具挤出的弹性体的重量。模具具有6.35mm的直径和0.5mm的厚度并且设置在直径为52mm的空心圆柱盘的底部的中心。
将预先以丸粒形式(2cm的厚度和52mm的直径)形成的40±4g弹性体放置在该设备中。将重量为1kg(±5g)的校准活塞放置在弹性体丸粒上。然后将组件放置在100℃±0.5℃的炉中。
由于在烘箱的第一个小时内条件不稳定,在t=1小时挤出的产品被切断,然后丢弃。
随后继续测量6小时±5分钟,在此期间产品留在烘箱中。在6小时结束时,将挤出产品样品切下,然后称重。测量结果是称重弹性体的重量。结果以相对于对照的基准100给出。数值越低,弹性体流动的阻力越大。
经改性的弹性体的实体分布的确定
使用的方法是本申请人名下的专利申请WO 2015/018599 A1中记载的建模方法,所述建模方法描述如下。
1-确定分批搅拌反应器中的官能化动力学模型的动力学常数(K)的比例的实施例
以实验的方式确定在链端被官能化的链、在链中部被官能化的链和星形支化(3个支链)的链的重量百分比以及粘度随(3-N,N-二甲基氨基丙基)三甲氧基硅烷/n-BuLi摩尔比的跳变
将91.6ml(70.5g)甲基环己烷、14.8ml(9.65g)丁二烯和0.49ml四氢化糠基醚在甲基环己烷中的0.078mol.l-1的溶液引入十一个250ml的玻璃瓶(Steinie瓶)。在中和了溶液(该溶液待加入正丁基锂(n-BuLi)以使其聚合)中的杂质后,加入1.90ml在甲基环己烷中的0.097mol.l-1的n-BuLi。在60℃下进行聚合。
15分钟之后,单体的转化程度达到95%。通过称重在200毫米汞柱的减压下在140℃下干燥的提取物确定该含量。使用相对于锂过量的甲醇使对照瓶(1号瓶)的反应停止。测量的“初始”固有粘度为0.66dl.g-1。将0.88ml(3-N,N-二甲基氨基丙基)三甲氧基硅烷在甲基环己烷中的0.1mol.l-1的溶液加入瓶2至9中存在的活性聚合物溶液(相对于Li 0.48摩尔当量),向瓶10中加入0.73ml的相同溶液(相对于Li 0.40摩尔当量)并且向瓶11中加入1.83ml的相同溶液(相对于Li 1.0摩尔当量)。在60℃下反应15分钟之后,通过加入0.4份/100份弹性体(phr)的4,4’-亚甲基双(2,6-二(叔丁基)酚)和0.2份/100份弹性体(phr)的N-(1,3-二甲基丁基)-N’-苯基-对-苯二胺从而对溶液进行抗氧化处理。通过在氮气流中在60℃下减压干燥12小时从而从溶液中分离由此处理的聚合物。
“最终”固有粘度、粘度跳变(定义为“最终”固有粘度与“初始”固有粘度的比例)以及失活链(P)、在链端被官能化的链(PA)、在链中部被官能化的链(P2A)和星形支化的链(P3A)的重量百分比显示于下表1中。
表1–实体P+PA/P2A/P3A分布的变化以及粘度随(3-N,N-二甲基氨基丙基)三甲氧基硅烷/n-BuLi摩尔比的跳变
活性二烯弹性体根据如下反应机制而官能化:
其中
-A表示官能化剂,
-PLi表示活性弹性体链,
-PA表示在链端被官能化的弹性体,
-P2A表示偶联的弹性体,
-P3A表示具有三个支链的星形支化的弹性体,并且
-ki表示反应Ri的动力学常数,
根据如下速率定律进行:
其中
-k1、k2和k3分别为反应R1、R2和R3的动力学常数(用(m3/mol).s-1表示),
-[PLi]为活性链的浓度(用mol/m3表示),
-[A]为改性剂A的浓度(用mol/m3表示),
-[PA]为在链端被官能化的弹性体的浓度(用mol/m3表示),
-[P2A]为偶联弹性体的浓度(用mol/m3表示),
-[P3A]为具有三个支链的星形支化的弹性体的浓度(用mol/m3表示),
动力学常数的比例K定义为:大于1。
根据本领域技术人员引入完美搅拌的分批反应器模型的官能化动力学模型(bibliographie:Villermeaux,J;Génie de la réaction chimique;1993)使得能够确定不同实体的分布。此外,链可能在聚合和/或官能化步骤的过程中失活(P)。因此,最终产物为失活弹性体(P)、在链端被官能化的弹性体(PA)、在链中部被官能化的弹性体(P2A)和星形支化的弹性体(P3A)的混合物。
对于上表1的实验点,根据完美搅拌的分批反应器模型(其代表这些实验所使用的反应器)的说明书确定K=102±1的值。
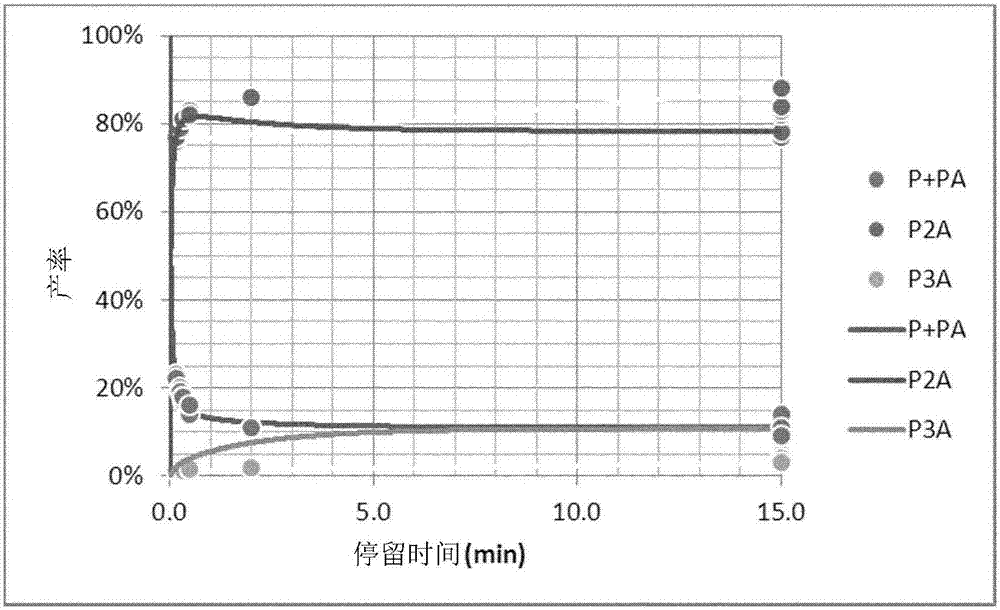
图1中显示了实体P、PA、P2A和P3A的分布随官能化剂/活性聚合物链(PLi)摩尔比的变化:模拟(线)和测量(点)。
2-确定分批搅拌反应器中的官能化动力学的实施例
以实验的方式确定在链端被官能化的链、在链中部被官能化的链和星形支化(3个支链)链的重量百分比随(3-N,N-二甲基氨基丙基)三甲氧基硅烷的偶联时间的变化(相对于Li~0.5摩尔当量)
将91.6ml(70.5g)甲基环己烷、14.8ml(9.65g)丁二烯和0.49ml四氢化糠基醚在甲基环己烷中的0.078mol.l-1的溶液引入二十二个250ml的玻璃瓶(Steinie瓶)。在中和了溶液(该溶液待加入正丁基锂(n-BuLi)以使其聚合)中的杂质后,加入1.90ml在甲基环己烷中的0.097mol.l-1的n-BuLi。在60℃下进行聚合。
15分钟之后,单体的转化程度达到95%。通过称重在200毫米汞柱的减压下在140℃下干燥的提取物确定该含量。将0.88ml(3-N,N-二甲基氨基丙基)三甲氧基硅烷在甲基环己烷中的0.1mol.l-1的溶液加入剩余二十一个瓶中存在的活性聚合物溶液(相对于Li 0.48摩尔当量)。在60℃下反应10秒(瓶12、13和14)、15秒(瓶15、16和17)、20秒(瓶18、19和20)、30秒(瓶21和22)、2分钟(瓶23)和15分钟(瓶24、25、26、27、28、29、30、31和32)之后,通过加入0.4份/100份弹性体(phr)的4,4’-亚甲基双(2,6-二(叔丁基)酚)和0.2份/100份弹性体(phr)N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对溶液进行抗氧化处理。通过在氮气流中在60℃下减压干燥12小时从而从溶液中分离由此处理的聚合物。
下表2显示了失活链(P)、在链端被官能化的链(PA)、在链中部被官能化的链(P2A)和星形支化的链(P3A)的重量百分比。
表2–实体P+PA/P2A/P3A的分布随(3-N,N-二甲基氨基丙基)三甲氧基硅烷的反应时间的变化
通过使用与之前实施例相同的动力学模型和K=102±1的值,确定动力学模型中的k1[PLi]值为104±0.2。图2中对比了完美搅拌的分批反应器中的模拟产率和测量产率随反应时间的变化。
3-确定连续构造中的官能化的动力学常数(K)的比例的实施例
在连续搅拌的聚合反应器(假设完美搅拌)的出口处将可变量的官能化剂注入连续中试聚合装置中,从而表征连续官能化工段。官能化工段由包括36个4升元件的Kenics型静态混合器和体积为32.5升的连续搅拌反应器(假设完美搅拌)组成。搅拌反应器中的最小停留时间为20分钟。
根据如下比例连续引入甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚:丁二烯的重量流速=2.85kg.h-1,苯乙烯的重量流速=1.25kg.h-1,单体的重量浓度=11重量%,60ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚连续引入32.5升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过线路入口中存在的不同成分引入的质子杂质。在反应器入口引入850μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为40分钟。保持温度在90℃。
在从反应器出口提取的样品上测量的转化程度为92.6%。
在聚合反应器的出口处以不同的量(不同的(3-N,N-二甲基氨基丙基)三甲氧基硅烷/PLi摩尔比)向活性聚合物溶液中加入(3-N,N-二甲基氨基丙基)三甲氧基硅烷在甲基环己烷中的溶液从而表征官能化过程。在由36个Kenics KMR型混合元件组成的静态混合器中混合该溶液然后使该溶液穿过空管(管中的总停留时间为3分钟(静态混合器空管)),穿过32.5升连续搅拌反应器(本领域技术人员假设完美搅拌)并且停留时间为40分钟。然后,加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺对聚合物进行抗氧化处理。
通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
图3中显示了测得的固有粘度跳变(SV)。
根据本领域技术人员,将上述官能化动力学模型并入管状反应器模型(Kenics静态混合器的代表),然后是完美搅拌的连续反应器(搅拌官能化反应器的代表)(bibliographie:Villermeaux,J;Génie de la réaction chimique;1993)并且能够确定PLi、P、PA、P2A和P3A实体的分布。
为了在通过官能化动力学模型计算的实体PLi、P、PA、P2A和P3A的分布和官能化前后的固有粘度的实验比例(SV)之间建立联系,理论上通过如下方程计算SV:
其中:
wPiA为实体PiA、PLi和P的重量份数;
Mw为重均分子量;
a为MHS(Mark-Houwink-Sakurada)方程的参数并且等于0.75;
gi’为星形支化聚合物的修正,例如:
其中:
b等于0.58(功:熔融聚合物的结构和流变学)。
假设停留时间足够长并且可以被视为无限长,通过最小化实验SV和计算SV之间的差值确定动力学常数K的比例。如图3所示,K值为101±1。
通过并入管状且完美搅拌的连续反应器模型的动力学模型计算的实体分布确定计算SV(图4)。
动态性质:
根据标准ASTM D 5992-96,在粘度分析仪(Metravib VA4000)上测量动力学性质特别是tanδmax。根据标准ASTM D 1349-99,记录在标准温度条件(23℃)下以10Hz频率经受简单交变正弦剪切应力的硫化组合物样品(厚度为2mm并且横截面积为79mm2的圆柱状试样)的响应。从0.1%至50%峰至峰(向外循环)然后从50%至0.1%峰至峰(返回循环)应变振幅扫描。更特别使用的结果是损耗因数tanδ。对于返回循环,标明观察的tanδ的最大值,记为tanδmax。该值代表材料的滞后性并且在该情况下代表滚动阻力:tanδmax的值越小,滚动阻力越低。在实施例中,以基数100给出动力学性质的结果。该值越高,滞后性越高。两个值之间2个点的差值被认为是明显的。
磨损
根据标准NF ISO 4659的说明使用Zwick磨损仪进行磨损质量损失的测量,其中在10N的接触压力下使圆柱形样本经受附接至转鼓表面的P60微粒的磨网的作用达40m的进程。测得的值为基材在磨损之后的体积损失(单位为mm3),所述值越小,抗磨性越好。相对于对照的基数100给出结果。在该情况下,该值代表胎面包含所述材料的轮胎的耐磨性;所述值越小,材料的抗磨性越好,因此胎面包含该材料的轮胎的耐磨性越好。
弹性体制备实施例
聚合物A的制备:SBR氨基烷氧基硅烷–在链中部官能化-根据本发明Tg-88℃
根据如下比例连续引入甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚:丁二烯的重量流速=4.013kg.h-1,苯乙烯的重量流速=0.122kg.h-1,单体的重量浓度=9.75重量%,15ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚连续引入32升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过第一反应器入口中存在的不同成分引入的质子杂质。在反应器入口引入850μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为35分钟。保持温度在95℃。
在聚合反应器的出口取出聚合物溶液的样品。加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。测量的“初始”固有粘度为1.98dl.g-1。通过SEC技术确定的数均分子量Mn为90 000g.mol-1并且多分散指数Ip为1.90。
在聚合反应器的出口处向活性聚合物溶液中加入440μmol/100g单体的(3-N,N-二甲基氨基丙基)三甲氧基硅烷(偶联和星形支化剂AdC)在甲基环己烷中的溶液(AdC/Li=0.52)。
在由36个Kenics KMR型混合元件组成的静态混合器构成的活塞型系统混合该溶液30秒然后使溶液穿过空管。然后在假设完美搅拌的反应器中混合该溶液35分钟,这些反应器(活塞型系统+假设完美搅拌的混合器)中的温度为95℃。
加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。
然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
测量的“最终”固有粘度为2.52dl.g-1。在该例子中,粘度跳变(其定义为所述“最终”粘度与所述“起始”粘度之比)为1.27。
该聚合物A的门尼粘度为70。
通过SEC技术确定的数均分子量Mn为168600g.mol-1并且多分散指数Ip为1.68。
通过NIR方法确定该聚合物的微观结构。相对于丁二烯单元,1,2-单元的含量为12.7%。苯乙烯的重量含量为2.1%。
该聚合物的玻璃化转变温度为-88℃。
通过上述建模方法获得官能化之后的实体分布:86%为官能化链(其中77%在链中部官能化)并且14%为星形支化的链。
聚合物B的制备:BR氨基烷氧基硅烷–在链中部官能化-根据本发明Tg-91℃
根据如下比例连续引入甲基环己烷、丁二烯和四氢化糠基乙醚:丁二烯的重量流速=4.135kg.h-1,单体的重量浓度=9.75重量%,15ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚连续引入32升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过第一反应器入口中存在的不同成分引入的质子杂质。在反应器入口引入850μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为35分钟。保持温度在95℃。
在聚合反应器的出口取出聚合物溶液的样品。加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。测量的“初始”固有粘度为1.97dl.g-1。通过SEC技术确定的数均分子量Mn为88000g.mol-1并且多分散指数Ip为1.90。
在聚合反应器的出口处向活性聚合物溶液中加入442μmol/100g单体的(3-N,N-二甲基氨基丙基)三甲氧基硅烷(偶联和星形支化剂AdC)在甲基环己烷中的溶液(AdC/Li=0.52)。
在由36个Kenics KMR型混合元件组成的静态混合器构成的活塞型系统混合该溶液30秒然后使溶液穿过空管。然后在假设完美搅拌的反应器中混合该溶液35分钟,这些反应器(活塞型系统+假设完美搅拌的混合器)中的温度为95℃。
加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。
然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
测量的“最终”固有粘度为2.54dl.g-1。在该例子中,粘度跳变(其定义为所述“最终”粘度与所述“起始”粘度之比)为1.27。
该聚合物B的门尼粘度为69。
通过SEC技术确定的数均分子量Mn为170000g.mol-1并且多分散指数Ip为1.70。
通过NIR方法确定该聚合物的微观结构。相对于丁二烯单元,1,2-单元的含量为12.5%。
该聚合物的玻璃化转变温度为-91℃。
通过上述建模方法获得官能化之后的实体分布:86%为官能化链(其中77%在链中部官能化)并且14%为星形支化的链。
聚合物C的制备:SBR氨基烷氧基硅烷–在链中部官能化-对比Tg-49℃
根据如下比例连续引入甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚:丁二烯的重量流速=2.874kg.h-1,苯乙烯的重量流速=1.204kg.h-1,单体的重量浓度=11重量%,60ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚连续引入32升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过第一反应器入口中存在的不同成分引入的质子杂质。在反应器入口引入870μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为40分钟。保持温度在90℃。
在聚合反应器的出口取出聚合物溶液的样品。加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。测量的“初始”固有粘度为1.66dl.g-1。通过SEC技术确定的数均分子量Mn为94000g.mol-1并且多分散指数Ip为1.90。
在聚合反应器的出口处向活性聚合物溶液中加入452μmol/100g单体的(3-N,N-二甲基氨基丙基)三甲氧基硅烷(偶联和星形支化剂AdC)在甲基环己烷中的溶液(AdC/Li=0.52)。
在由36个Kenics KMR型混合元件组成的静态混合器构成的活塞型系统混合该溶液30秒然后使溶液穿过空管。然后在假设完美搅拌的反应器中混合该溶液35分钟,这些反应器(活塞型系统+假设完美搅拌的混合器)中的温度为95℃。
加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。
然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
测量的“最终”固有粘度为2.1dl.g-1。在该例子中,粘度跳变(其定义为所述“最终”粘度与所述“起始”粘度之比)为1.30。
该聚合物C的门尼粘度为67。
通过SEC技术确定的数均分子量Mn为141200g.mol-1并且多分散指数Ip为1.8。
通过NIR方法确定该聚合物的微观结构。相对于丁二烯单元,1,2-单元的含量为24.2%。苯乙烯的重量含量为26.4%。
该聚合物的玻璃化转变温度为-49℃。
通过上述建模方法获得官能化之后的实体分布:86%为官能化链(其中77%在链中部官能化)并且14%为星形支化的链。
聚合物D的制备:SBR氨基烷氧基硅烷–在链中部官能化-对比Tg-88℃
根据如下比例连续引入甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚:丁二烯的重量流速=4.013kg.h-1,苯乙烯的重量流速=0.122kg.h-1,单体的重量浓度=9.75重量%,15ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚连续引入32升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过第一反应器入口中存在的不同成分引入的质子杂质。在反应器入口引入850μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为35分钟。保持温度在95℃。
在聚合反应器的出口取出聚合物溶液的样品。加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。测量的“初始”固有粘度为1.98dl.g-1。通过SEC技术确定的数均分子量Mn为90000g.mol-1并且多分散指数Ip为1.90。
在聚合反应器的出口处向活性聚合物溶液中加入380μmol/100g单体的(3-N,N-二甲基氨基丙基)三甲氧基硅烷(偶联和星形支化剂AdC)在甲基环己烷中的溶液(AdC/Li=0.45)。
在由36个Kenics KMR型混合元件组成的静态混合器构成的活塞型系统混合该溶液30秒然后使溶液穿过空管。然后在假设完美搅拌的反应器中混合该溶液35分钟,这些反应器(活塞型系统+假设完美搅拌的混合器)中的温度为95℃。
加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。
然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
测量的“最终”固有粘度为2.67dl.g-1。在该例子中,粘度跳变(其定义为所述“最终”粘度与所述“起始”粘度之比)为1.35。
该聚合物D的门尼粘度为77。
通过SEC技术确定的数均分子量Mn为165000g.mol-1并且多分散指数Ip为1.70。
通过NIR方法确定该聚合物的微观结构。相对于丁二烯单元,1,2-单元的含量为12.7%。苯乙烯的重量含量为2.1%。
该聚合物的玻璃化转变温度为-88℃。
通过上述建模方法获得官能化之后的实体分布:67%为在链中部官能化的链并且33%为星形支化的链。
聚合物E的制备:SBR氨基烷氧基硅烷–在链中部官能化-根据本发明Tg-88℃
根据如下比例连续引入甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚:丁二烯的重量流速=4.013kg.h-1,苯乙烯的重量流速=0.122kg.h-1,单体的重量浓度=9.75重量%,15ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚连续引入32升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过第一反应器入口中存在的不同成分引入的质子杂质。在反应器入口引入850μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为35分钟。保持温度在95℃。
在聚合反应器的出口取出聚合物溶液的样品。加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。测量的“初始”固有粘度为1.98dl.g-1。通过SEC技术确定的数均分子量Mn为90000g.mol-1并且多分散指数Ip为1.90。
在聚合反应器的出口处向活性聚合物溶液中加入425μmol/100g单体的(3-N,N-二甲基氨基丙基)三甲氧基硅烷(偶联和星形支化剂AdC)在甲基环己烷中的溶液(AdC/Li=0.50)。
在由36个Kenics KMR型混合元件组成的静态混合器构成的活塞型系统混合该溶液30秒然后使溶液穿过空管。然后在假设完美搅拌的反应器中混合该溶液35分钟,这些反应器(活塞型系统+假设完美搅拌的混合器)中的温度为95℃。
加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。
然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
测量的“最终”固有粘度为2.54dl.g-1。在该例子中,粘度跳变(其定义为所述“最终”粘度与所述“起始”粘度之比)为1.30。
该聚合物E的门尼粘度为71。
通过SEC技术确定的数均分子量Mn为165 000g.mol-1并且多分散指数Ip为1.70。
通过NIR方法确定该聚合物的微观结构。相对于丁二烯单元,1,2-单元的含量为12.7%。苯乙烯的重量含量为2.1%。
该聚合物的玻璃化转变温度为-88℃。
通过上述建模方法获得官能化之后的实体分布:83%为官能化链(其中76%在链中部官能化)并且18%为星形支化的链。
聚合物F的制备:SBR环氧化物+烷氧基硅烷–在链中部官能化-根据本发明Tg-88℃
根据如下比例连续引入甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚:丁二烯的重量流速=4.013kg.h-1,苯乙烯的重量流速=0.122kg.h-1,单体的重量浓度=9.75重量%,15ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚连续引入32升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过第一反应器入口中存在的不同成分引入的质子杂质。在反应器入口引入850μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为35分钟。保持温度在95℃。
在聚合反应器的出口取出聚合物溶液的样品。加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。测量的“初始”固有粘度为1.95dl.g-1。通过SEC技术确定的数均分子量Mn为88 000g.mol-1并且多分散指数Ip为1.89。
在聚合反应器的出口处向活性聚合物溶液中加入440μmol/100g单体的(3-环氧丙氧基丙基)三甲氧基硅烷(偶联和星形支化剂AdC)在甲基环己烷中的溶液(AdC/Li=0.52)。
在由36个Kenics KMR型混合元件组成的静态混合器构成的活塞型系统混合该溶液30秒然后使溶液穿过空管。然后在假设完美搅拌的反应器中混合该溶液35分钟,这些反应器(活塞型系统+假设完美搅拌的混合器)中的温度为95℃。
加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。
然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
测量的“最终”固有粘度为2.50dl.g-1。在该例子中,粘度跳变(其定义为所述“最终”粘度与所述“起始”粘度之比)为1.28。
该聚合物F的门尼粘度为71。
通过SEC技术确定的数均分子量Mn为170 200g.mol-1并且多分散指数Ip为1.66。
通过NIR方法确定该聚合物的微观结构。相对于丁二烯单元,1,2-单元的含量为12.7%。苯乙烯的重量含量为2.1%。
该聚合物的玻璃化转变温度为-88℃。
通过上述建模方法获得官能化之后的实体分布:83%为官能化链(其中79%在链中部官能化)并且17%为星形支化的链。
聚合物G的制备:BR环氧化物+烷氧基硅烷–在链中部官能化-根据本发明Tg-91℃
根据如下比例连续引入甲基环己烷、丁二烯和四氢化糠基乙醚:丁二烯的重量流速=4.135kg.h-1,单体的重量浓度=9.75重量%,15ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、和四氢化糠基乙醚连续引入32升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过第一反应器入口中存在的不同成分引入的质子杂质。在反应器入口引入850μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为35分钟。保持温度在95℃。
在聚合反应器的出口取出聚合物溶液的样品。加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。测量的“初始”固有粘度为1.99dl.g-1。通过SEC技术确定的数均分子量Mn为90000g.mol-1并且多分散指数Ip为1.91。
在聚合反应器的出口处向活性聚合物溶液中加入440μmol/100g单体的(3-环氧丙氧基丙基)三甲氧基硅烷(偶联和星形支化剂AdC)在甲基环己烷中的溶液(AdC/Li=0.52)。
在由36个Kenics KMR型混合元件组成的静态混合器构成的活塞型系统混合该溶液30秒然后使溶液穿过空管。然后在假设完美搅拌的反应器中混合该溶液35分钟,这些反应器(活塞型系统+假设完美搅拌的混合器)中的温度为95℃。
加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。
然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
测量的“最终”固有粘度为2.55dl.g-1。在该例子中,粘度跳变(其定义为所述“最终”粘度与所述“起始”粘度之比)为1.28。
该聚合物G的门尼粘度为70。
通过SEC技术确定的数均分子量Mn为173000g.mol-1并且多分散指数Ip为1.71。
通过NIR方法确定该聚合物的微观结构。相对于丁二烯单元,1,2-单元的含量为12.5%。
该聚合物的玻璃化转变温度为-91℃。
通过上述建模方法获得官能化之后的实体分布:85%为官能化链(其中76%在链中部官能化)并且15%为星形支化的链。
聚合物H的制备:SBR环氧化物+烷氧基硅烷–在链中部官能化-对比Tg-48℃
根据如下比例连续引入甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚:丁二烯的重量流速=2.874kg.h-1,苯乙烯的重量流速=1.204kg.h-1,单体的重量浓度=11重量%,60ppm的四氢化糠基乙醚,将甲基环己烷、丁二烯、苯乙烯和四氢化糠基乙醚连续引入32升连续搅拌反应器(本领域技术人员假设完美搅拌)。引入足够量的正丁基锂(n-BuLi)从而中和通过第一反应器入口中存在的不同成分引入的质子杂质。在反应器入口引入870μmol n-BuLi/100g单体。
计算不同的流速使得反应器中的平均停留时间为40分钟。保持温度在90℃。
在聚合反应器的出口取出聚合物溶液的样品。加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。测量的“初始”固有粘度为1.65dl.g-1。通过SEC技术确定的数均分子量Mn为92000g.mol-1并且多分散指数Ip为1.90。
在聚合反应器的出口处向活性聚合物溶液中加入450μmol/100g单体的(3-环氧丙氧基丙基)三甲氧基硅烷(偶联和星形支化剂AdC)在甲基环己烷中的溶液(AdC/Li=0.52)。
在由36个Kenics KMR型混合元件组成的静态混合器构成的活塞型系统混合该溶液30秒然后使溶液穿过空管。然后在假设完美搅拌的反应器中混合该溶液35分钟,这些反应器(活塞型系统+假设完美搅拌的混合器)中的温度为95℃。
加入0.4phr的2,2’-亚甲基双(4-甲基-6-(叔丁基)酚)和0.2phr的N-(1,3-二甲基丁基)-N’-苯基-对苯二胺从而对由此获得的聚合物进行抗氧化处理。
然后通过蒸汽汽提操作从溶液中分离由此处理的聚合物,然后在100℃开炼机上干燥。
测量的“最终”固有粘度为2.14dl.g-1。在该例子中,粘度跳变(其定义为所述“最终”粘度与所述“起始”粘度之比)为1.30。
该聚合物H的门尼粘度为68。
通过SEC技术确定的数均分子量Mn为143000g.mol-1并且多分散指数Ip为1.75。
通过NIR方法确定该聚合物的微观结构。相对于丁二烯单元,1,2-单元的含量为24.0%。苯乙烯的重量含量为27.0%。
该聚合物的玻璃化转变温度为-48℃。
通过上述建模方法获得官能化之后的实体分布:85%为官能化链(其中77%在链中部官能化)并且15%为星形支化的链。
聚合物I的制备:BR巯基烷氧基硅烷-链中部官能化-根据本发明Tg-91℃
将7.5kg丁二烯和313ml四氢呋喃在甲基环己烷中的0.349mol.l-1的溶液注入90升反应器中,所述反应器保持在约2bar的氮压下并且包含43.9kg甲基环己烷。在中和了溶液(该溶液待加入正丁基锂以使其聚合)中的杂质后,加入1043ml在甲基环己烷中的0.063mol.l-1的正丁基锂。在50℃下进行聚合。
95分钟之后,单体的转化程度达到89%。通过称重在200毫米汞柱的减压下在140℃下干燥的提取物确定该含量。将528ml 3,3-甲氧基-8,8,9,9-四甲基-2-氧杂-7-硫代-3,8-二硅杂癸烷在甲基环己烷中的0.020mol.l-1的溶液加入活性聚合物的溶液中。在50℃下反应30分钟之后,然后将528ml 3,3-甲氧基-8,8,9,9-四甲基-2-氧杂-7-硫代-3,8-二硅杂癸烷在甲基环己烷中的0.030mol.l-1的溶液加入该聚合物溶液中。在50℃下反应30分钟之后,通过加入0.8份/100份弹性体(phr)的4,4’-亚甲基双(2,6-二(叔丁基)酚)和0.2份/100份弹性体(phr)的N-(1,3-二甲基丁基)-N’-苯基-对-苯二胺从而对溶液进行抗氧化处理。通过蒸汽汽提操作从溶液中分离由此处理的共聚物,然后在100℃开炼机上干燥。
该共聚物的微观结构通过NIR方法加以确定:1,4-反式单元的重量含量为49%,1,4-顺式单元的重量含量为37%并且1,2-单元的重量含量为14%。
该共聚物的玻璃化转变温度为-91℃。
聚合物E的门尼粘度为42。
通过高分辨率SEC方法获得官能化之后的实体分布:70%为在链中部官能化的链,16%为非官能化链并且14%为星形支化的链。
聚合物J的制备:SBR巯基烷氧基硅烷–在链中部官能化-对比Tg-48℃
将3.0kg苯乙烯和4.7kg丁二烯以及660ml四氢呋喃在甲基环己烷中的0.59mol.l-1的溶液注入90升反应器中,所述反应器保持在约2bar的氮压下并且包含44.3kg甲基环己烷。在中和了溶液(该溶液待加入正丁基锂以使其聚合)中的杂质后,加入825ml在甲基环己烷中的0.063mol.l-1的正丁基锂。在50℃下进行聚合。
45分钟之后,单体的转化程度达到69%。通过称重在200毫米汞柱的减压下在140℃下干燥的提取物确定该含量。将412ml 3,3-甲氧基-8,8,9,9-四甲基-2-氧杂-7-硫代-3,8-二硅杂癸烷在甲基环己烷中的0.020mol.l-1的溶液加入活性聚合物的溶液中。在50℃下反应30分钟之后,然后将412ml 3,3-甲氧基-8,8,9,9-四甲基-2-氧杂-7-硫代-3,8-二硅杂癸烷在甲基环己烷中的0.030mol.l-1的溶液加入该聚合物溶液中。在50℃下反应30分钟之后,通过加入0.8份/100份弹性体(phr)的4,4’-亚甲基双(2,6-二(叔丁基)酚)和0.2份/100份弹性体(phr)的N-(1,3-二甲基丁基)-N’-苯基-对-苯二胺从而对溶液进行抗氧化处理。通过蒸汽汽提操作从溶液中分离由此处理的共聚物,然后在100℃开炼机上干燥。
该共聚物的微观结构通过NIR方法加以确定:1,4-反式单元的重量含量为48%,1,4-顺式单元的重量含量为28%,并且1,2-单元的重量含量为24%,这三个含量中的每个含量涉及丁二烯单元。苯乙烯的重量含量为27.5%。
该共聚物的玻璃化转变温度为-48℃。
聚合物J的门尼粘度为66。
通过高分辨率SEC方法获得官能化之后的实体分布:70%为在链中部官能化的链,16%为非官能化链并且14%为非官能化的星形支化的链。
聚合物K的制备:BR巯基烷氧基硅烷–在链中部官能化-对比Tg-91℃
将7.5kg丁二烯和313ml四氢呋喃在甲基环己烷中的0.349mol.l-1的溶液注入90升反应器中,所述反应器保持在约2bar的氮压下并且包含43.9kg甲基环己烷。在中和了溶液(该溶液待加入正丁基锂以使其聚合)中的杂质后,加入1019ml在甲基环己烷中的0.063mol.l-1的正丁基锂。在50℃下进行聚合。
95分钟之后,单体的转化程度达到88%。通过称重在200毫米汞柱的减压下在140℃下干燥的提取物确定该含量。将516ml MeSiCl3在甲基环己烷中的0.015mol.l-1的溶液加入活性聚合物的溶液中。在50℃下反应15分钟之后,然后将590ml 3,3-甲氧基-8,8,9,9-四甲基-2-氧杂-7-硫代-3,8-二硅杂癸烷在甲基环己烷中的0.035mol.l-1的溶液加入该聚合物溶液中。在50℃下反应30分钟之后,通过加入0.8份/100份弹性体(phr)的4,4’-亚甲基双(2,6-二(叔丁基)酚)和0.2份/100份弹性体(phr)的N-(1,3-二甲基丁基)-N’-苯基-对-苯二胺从而对溶液进行抗氧化处理。通过蒸汽汽提操作从溶液中分离由此处理的共聚物,然后在100℃开炼机上干燥。
该共聚物的微观结构通过NIR方法加以确定:1,4-反式单元的重量含量为49%,1,4-顺式单元的重量含量为36%并且1,2-单元的重量含量为15%。
该共聚物的玻璃化转变温度为-90℃。
聚合物K的门尼粘度为61。
通过高分辨率SEC方法获得官能化之后的实体分布:51%为在链中部官能化的链,15%为非官能化链并且34%为星形支化的链。
橡胶组合物:
使用弹性体A至K制备胎面型橡胶组合物,每种橡胶组合物包含二氧化硅作为增强填料。
在第一步骤中通过热机械加工,接着在第二精制步骤中通过机械加工制备如下组合物的每一个。
将弹性体、三分之二的二氧化硅、炭黑、偶联剂和油以及(约一分钟之后)剩余的增强填料、树脂、抗氧化剂、硬脂酸和抗臭氧蜡以及(约两分钟之后)氧化锌连续引入Banbury型实验室密炼机中,所述密炼机的容量为400cm3并且填充至70%并且初始温度为90℃。
热机械加工的步骤进行4至5分钟直至大约160℃的最大出料温度。
由此进行热机械加工的上述第一步骤,需要指出的是在该第一步骤中叶片的平均速度为50rpm。
回收由此获得的混合物,接着进行冷却,在30℃下向开炼机(均匀修整器)中加入硫和促进剂,使得组合的混合物进一步混合3至4分钟的时间(机械加工的上述第二步骤)。
接着,将由此获得的组合物压延成橡胶板材(厚度为2至3毫米)或薄片材的形式,以测量它们的物理或机械性质,或者压延成在切割和/或组装成所需的尺寸之后可直接使用的成型件形式,例如作为轮胎半成品,特别是用于胎面。
交联在150℃下进行40分钟。
每种组合物具有如下配方(以phr表示:份/100份弹性体):
二氧化硅:来自Solvay的Z1165MP,BET表面积约160m2/g
炭黑:来自Cabot Corporation的ASTM级N234
油:来自Novance的Lubrirob Tod 1880
树脂:来自ExxonMobil的ECR-373
偶联剂:来自Degussa的TESPT Si69
促进剂:来自Flexsys的Santocure CBS
抗氧化剂:来自Flexsys的6-对苯二胺(6PPD)
结果:
下表1至3显示的结果表明,使用根据本发明的聚合物能够实现磨损的明显改进和滚动阻力的改进,实现聚合物的良好水平的抗流动性。
表1:
相对于样本C的基数100进行标准化
表2:
相对于样本H的基数100进行标准化
表3:
相对于样本J的基数100进行标准化。
- 还没有人留言评论。精彩留言会获得点赞!
- 包含/包括来自四氟乙烯和具有官能团和可聚合碳-碳双键的单体的重复单元的不可熔融 ...的制作方法
- 包含来自四氟乙烯和具有官能团和可聚合碳-碳双键的烃单体的重复单元的可熔融流动 ...的制作方法
- 具有官能团的乙烯/四氟乙烯共聚物的反应方法
- 含有苯乙烯官能团的氧杂环丁烷化合物的制作方法
- 单官能团8-羟基喹啉金属配合物的烯类单体及其制法和用途的制作方法
- 多官能团8-羟基喹啉金属配合物的烯类单体及其制法和用途的制作方法
- 使用隐色染料在镜片上生成可见标记的方法
- 包括官能团部分的聚(乙烯醇)-共-聚(乙烯胺)聚合物的制作方法
- 包含官能化水溶性聚合物层的纤维基载体,及其生产方法和用途
- 含有羧酸端官能团的聚乙烯和聚乙烯混合物的防晒组合物的制作方法