一种耐盐型阴离子交换膜色谱介质及其制备方法和用途与流程
本发明属于分离膜领域,具体涉及一种膜分离介质及其制备方法和用途,尤其涉及一种耐盐型阴离子交换膜色谱介质及其制备方法和用途。
背景技术:
膜色谱是一种膜孔道效应和色谱选择性吸附相结合的技术。由于膜内的传质是对流形式,因此膜色谱在操作速度上有明显的优势,同时床层薄,处理量较大,它的纯化操作时间远远小于柱层析(以扩散传质为主)。因此,膜色谱能够更高效地生产生物药物(蛋白类、疫苗类以及多肽类药物),同时温和的处理条件能更好的保持蛋白活性,满足现阶段市场日益增长地对生物类药物的需求。
耐盐型阴离子交换膜色谱介质一般带有含较多氨基的配基,其表面密集的电荷使其能够耐受高盐体系下的静电屏蔽作用。因此,这种分离介质能够在较高的离子强度下纯化目标生物分子,能够免除传统离子交换柱层析或膜色谱的前处理步骤(超滤换液、稀释以降低离子强度),大大提高了生产效率,节约材料和人力成本。制备耐盐型阴离子交换膜色谱介质的底膜材料一般为再生纤维素,其机械性能较差、容易降解、物理化学稳定性较差(重复使用差,通常为一次性使用)。同时,由一些惰性材料(如聚偏二氟乙烯、聚四氟乙烯、磺化聚砜等)制成的有机膜,虽然机械性能较好,但难以活化(用于配基偶联),亲水性也较差(易产生膜污染)。
如cn1768893a公开了一种以聚砜为基膜的螯合型中空纤维亲和膜色谱介质的制造方法,该法采用付氏反应成功地对基膜进行了活化改性,并偶联了螯合配基。但是,活化阶段需使用催化剂和有机溶剂,提高了制备成本,同时膜面容易受损,制备工艺较为复杂。cn1203363a公开了一种以木纤维素复合膜作为基质,然后固载金属离子、三嗪染料或蛋白配基制备亲和膜色谱的制造方法,该法首先需对膜进行酸处理产生羟基,接着通过高碘酸氧化形成醛基,最后偶联配基。然而三步反应过程较为复杂,同时酸化氧化反应会破坏膜表面结构,降低机械性能。另外,也有报道在惰性膜材料上接枝亲水性物质,从而改善膜的亲水性,然后再功能化偶联配基制备膜色谱介质的报道(gan,h.y.;shang,z.h.etal.,newaffinitynylonmembraneusedforadsorptionofgamma-globulin,j.chromatogr.a,2000(867)161-168.),但这些方法同样较为复杂,反应难以控制,另外亲水性物质能否均匀覆盖在整个膜上存在问题。
多巴胺是人体内分泌的一种重要的激素和神经递质体,一般情况下,它可以在碱性条件和氧气存在的环境中发生氧化自聚反应,形成的聚多巴胺层可以牢固地粘附于各种基材,它也被称为“生物胶水”。由于聚多巴胺中的儿茶酚基团能够通过迈克尔加成反应和席夫碱反应偶联带氨基或巯基的化合物,因此,这种方法可以有效地对有机/无机膜进行功能化改性。
如cn104028110a公开了一种利用聚多巴胺改善正渗透膜筛网的亲水性,多巴胺沉积使得改性后的渗透膜具有更好的渗透性和抗污染性能。因此,通过多巴胺氧化自聚在膜面形成聚多巴胺层,可以提高有机/无机膜的亲水性,同时聚多巴胺上的儿茶酚通过与耐盐型阴离子交换配基分子反应,构建以聚多巴胺为中间功能层的膜色谱。同时,聚多巴胺层可以提高膜材料的机械性能,使得所制备的膜色谱介质可以在高流速下操作。该制备方案均在水溶液中进行,制备过程温和、安全、绿色。
然而,目前并没有相关文献资料利用聚多巴胺改善有机/无机膜的亲水性,同时偶联耐盐型阴离子交换配基进行生物药物分离的报道。另一方面,在生物药物生产领域,为了提高生产效率,有待开发一种更高效的分离介质。
技术实现要素:
针对现有技术的不足,本发明的目的在于提供一种耐盐型(处理较高离子强度的料液)阴离子交换膜色谱介质,本发明提供的膜色谱介质具有耐高盐、高蛋白选择性和可在高流速下操作等特点。
为达到此发明目的,本发明采用以下技术方案:
一种耐盐型阴离子交换膜色谱分离介质,所述分离介质含有粘附在微滤膜内的聚多巴胺涂层,在所述聚多巴胺涂层上偶联耐盐型阴离子交换配基。
本发明的膜色谱介质不仅通过聚多巴胺粘附提高其机械强度,而且所含聚多巴胺涂层提高了膜面亲水性,降低了非特异性吸附和膜污染。另一方面,聚多巴胺也可以通过表面活性基团牢固地结合吸附性配基。本发明所述膜色谱分离介质能在较高离子强度、高流速下分离纯化生物药物分子。
在本发明中,微滤膜可以为商品化的均质膜或有机复合膜。
优选地,所述微滤膜的厚度为50-600微米,例如60微米、90微米、110微米、130微米、170微米、200微米、230微米、260微米、290微米、330微米、360微米、400微米、420微米、440微米、480微米或495微米、520微米、550微米、585微米等。
优选地,所述均质膜的厚度为90-200微米,例如为95微米、110微米、130微米、150微米、170微米、195微米等。
优选地,所述有机复合膜由分离层和支撑层组成。
优选地,所述有机复合膜的分离层厚度为0.1-40微米,例如0.1微米、0.5微米、0.8微米、1微米、1.2微米、1.8微米、2微米、2.5微米、5微米、8微米、10微米、15微米、18微米、24微米、27微米、30微米、32微米、37微米或39微米等,支撑层厚度为90-500微米,例如95微米、110微米、130微米、170微米、200微米、230微米、260微米、290微米、330微米、360微米、400微米、420微米、440微米、480微米或495微米等。
优选地,所述膜色谱介质的孔径为0.1-10微米,例如0.1微米、0.5微米、0.8微米、1微米、1.2微米、1.8微米、2微米、2.5微米、5微米、8微米或9微米等。
优选地,所述均质膜的材料为聚丙烯、磺化聚醚砜、聚偏二氟乙烯、聚四氟乙烯、磺化聚砜、醋酸纤维素、聚酰胺、聚醚砜或聚乙烯醇中的任意一种或至少两种的组合。
优选地,所述有机复合膜的分离层的材料为聚丙烯、磺化聚醚砜、聚偏二氟乙烯、聚四氟乙烯、磺化聚砜、醋酸纤维素、聚酰胺、聚哌嗪、聚醚砜或聚乙烯醇中的任意一种或至少两种的组合。
优选地,所述有机复合膜的支撑层的材料为聚烯烃如聚丙烯或聚酯中的任意一种或至少两种的组合。
优选地,所述聚多巴胺涂层是由多巴胺在碱性条件下自聚而形成。
优选地,所述耐盐型阴离子交换配基为胍基丁胺(agmatine)、三(2-氨乙基)胺(tris-2-aminoethylamine)、聚丙烯胺(polyallylamine)、聚乙烯亚胺(polyethyleneimine)或聚亚己基双胍(polyhexamethylenebiguanide)中的任意一种或至少两种的组合。
本发明的目的之一还在于提供一种本发明所述的耐盐型阴离子交换膜色谱分离介质的制备方法,包括以下步骤:
(1)将微滤膜浸没于碱性多巴胺溶液中,利用多巴胺氧化自聚的性能在微滤膜表面及孔内沉积一层纳米级厚度的聚多巴胺层;
(2)向步骤(1)得到的沉积有聚多巴胺的微滤膜上加入含有耐盐型阴离子交换配基的溶液,使所述配基与聚多巴胺进行偶联反应,得到耐盐型阴离子交换膜色谱。在加入含有耐盐型阴离子交换配基的溶液之前,优选将步骤(1)得到的沉积有聚多巴胺的微滤膜用去离子水洗涤。
作为优选,本发明的制备方法中,步骤(1)中所述多巴胺溶液为将多巴胺溶解于磷酸钠缓冲液、tris-hcl缓冲液或碳酸钠-碳酸氢钠缓冲液中得到的溶液,优选为将多巴胺溶解于tris-hcl缓冲液中得到的溶液。
优选地,所述多巴胺溶液的浓度为0.1-8.0g/l,例如0.2mg/ml、0.5mg/ml、1.0mg/ml、2.0mg/ml、3.0mg/ml、4.0mg/ml、5.0mg/ml、6.0mg/ml、7.0mg/ml或7.5mg/ml等。
优选地,所述多巴胺溶液的ph为7.2-10.5,例如7.5、8.0、8.5、9.0、9.5、10或10.1等。
优选地,所述多巴胺氧化自聚的环境为在空气存在下进行。
优选地,所述多巴胺氧化自聚在室温搅拌下进行,优选搅拌速度为20-400转/分钟,例如30转/分钟、60转/分钟、80转/分钟、90转/分钟、100转/分钟、120转/分钟、140转/分钟、160转/分钟、180转/分钟、240转/分钟、280转/分钟、320转/分钟、360转/分钟或390转/分钟等。
优选地,所述多巴胺氧化自聚的时间为1-36小时,例如2小时、3小时、5小时、7小时、9小时、10小时、15小时、18小时、21小时、25小时、28小时、32小时或35小时等。
作为优选,本发明的制备方法中,步骤(2)中所述配基溶液为配基溶于碳酸钠-碳酸氢钠缓冲液、磷酸缓冲液、水或tris-hcl缓冲液中得到的溶液,优选为将配基溶解于碳酸钠-碳酸氢钠缓冲液、磷酸缓冲溶液或tris-hcl缓冲溶液中得到的溶液。
优选地,所述配基溶液的浓度为0.1-4.0mg/ml,例如0.15g/l、0.3g/l、0.5g/l、0.8g/l、1g/l、1.3g/l、1.5g/l、1.8g/l、2g/l、2.4g/l、2.8g/l、3g/l、3.3g/l、3.5g/l、3.8g/l等,优选3.0mg/ml。
优选地,所述碳酸钠-碳酸氢钠缓冲液、磷酸缓冲溶液或tris-hcl缓冲溶液的ph值为5.0-11.0,例如5.2、5.6、5.8、6.0、6.4、6.8、7.0、7.2、7.4、7.8、8.0、8.2、8.5、8.8、9.0、9.3、9.5、10.0、10.5等,优选为5.5-9.5,进一步优选为9.0。
优选地,所述偶联反应的温度为15-80℃,例如17℃、20℃、30℃、40℃、50℃、60℃、70℃或78℃等,优选30℃。
优选地,所述偶联反应时搅拌速度为20-400转/分钟,例如30转/分钟、60转/分钟、80转/分钟、90转/分钟、100转/分钟、120转/分钟、140转/分钟、160转/分钟、180转/分钟、240转/分钟、280转/分钟、320转/分钟、360转/分钟或390转/分钟等。
优选地,所述配基与聚多巴胺偶联反应的时间为1-48小时,例如1.5小时、3小时、5小时、7小时、9小时、10小时、15小时、18小时、21小时、25小时、28小时、32小时、36小时、40小时、44小时或47小时等。
本发明的目的之一还在于提供本发明所述的耐盐型阴离子交换膜色谱介质在生物药物分离纯化中的用途,特别是在较高离子强度、高流速下分离纯化生物药物分子中的用途。
本发明所述的耐盐型阴离子交换膜色谱介质具有耐高盐、选择性高和耐高压等特点,使其对生物产品的处理效率较高。在150-300mm的盐浓度和40-100膜体积/分钟的流速下,通过聚多巴胺偶联的耐盐型阴离子交换配基能有效吸附带负电的生物分子,从而与带正电的生物分子进行分离。
优选地,所述生物药物为蛋白类、疫苗类或多肽类药物。
相对于现有技术,本发明具有以下有益效果:
本发明的膜色谱介质不仅保留了原高分子微滤膜较高的机械性能,同时通过聚多巴胺粘附进一步提高机械强度至少1.5倍以上,并且聚多巴胺提高了膜面亲水性,降低非特异性吸附和膜污染。另一方面,聚多巴胺可以牢固地结合吸附性配基。本发明的耐盐型阴离子交换膜色谱介质可用于生物药物(蛋白类、疫苗类以及多肽类药物)纯化。在高流速纯化药物的过程中,所述膜色谱介质的高亲水性和高机械强度可大大提高其承压能力。同时,整个制备过程高效、简易、成本低、绿色环保(不使用有机溶剂)。
附图说明
图1是本发明的耐盐型阴离子交换膜色谱介质的制备工艺流程示意图,其中1为微滤膜,2为聚多巴胺涂层,3为耐盐型配基分子;
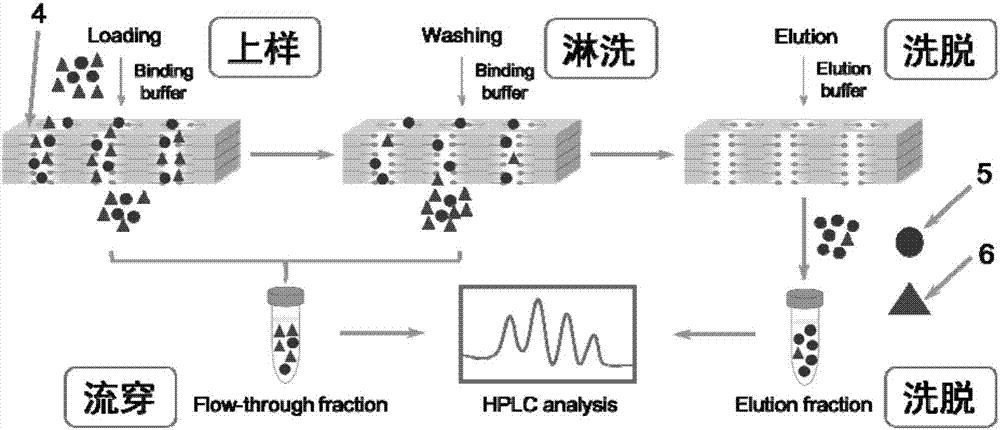
图2是本发明的耐盐型阴离子交换膜色谱介质进行生物分子纯化的示意图,其中4为耐盐型阴离子交换膜色谱介质,5为待纯化生物分子,6为杂质分子;
图3是实施例1制备的膜色谱介质应用时的层析图谱;
图4是实施例1制备的膜色谱介质应用时的分析结果;
图5是实施例2制备的膜色谱介质应用时的层析图谱;
图6是实施例2制备的膜色谱介质应用时的电泳图谱;
图7是实施例3制备的膜色谱介质应用时的层析图谱。
具体实施方式
为便于理解本发明,本发明列举实施例如下。本领域技术人员应该明了,所述实施例仅仅用于帮助理解本发明,不应视为对本发明的具体限制。
实施例1
在本实施例中,具备耐盐型阴离子交换膜色谱介质的制备方法如下(其制备工艺流程可以形象地以图1所示示意图来说明),具体包括以下步骤:
将160mg多巴胺分子加入20ml30mmtris-hcl缓冲液中(ph9.5),同时将一片孔径为2.0微米聚丙烯膜(图1中标识1)浸入多巴胺溶液中,接着在摇床中以200转/分钟的转速在常温下反应24小时。反应结束后,取出膜片,用去离子水冲洗三遍。然后,将经多巴胺涂覆的膜片(图1中标识2)浸入2mg/ml胍基丁胺和0.5mg/ml聚丙烯胺(图1中标识3)的tris-hcl缓冲溶液(ph=9.0,50ml),在摇床中以200转/分钟的转速反应24小时(30℃)。反应结束后,用去离子水冲洗膜片。最后用氮气吹干,保存。
应用上述制备的膜色谱介质对人免疫球蛋白(igg)和人血清白蛋白(hsa)进行分离纯化,其分离过程可以图2来说明,具体分离方法和结果如下:
首先,将新制备的膜色谱介质裁剪成直径为2.5cm的膜片,将八片膜堆叠(图2中标识4)放入容器中,使容器与蛋白层析系统相连接。待分离蛋白料液为10mgigg(图2中标识6)和2mghsa(图2中标识5)溶解于加有0.2m氯化钠的磷酸缓冲液(ph7.0,20mm)。上述料液进样至蛋白层析系统,料液流过膜色谱介质,接着用加有0.2m氯化钠的磷酸缓冲液(ph7.0,20mm)淋洗膜片,最后,与膜结合的蛋白用加有2m氯化钠的磷酸缓冲液(ph7.0,20mm)进行洗脱(图3为层析图谱)。收集淋洗流穿和洗脱液(图4为分析结果),进行蛋白回收率和纯度的分析。
纯化结果为该耐压型膜色谱介质能够在0.2m氯化钠的高盐体系下对igg和hsa进行有效分离,获得igg和hsa的回收率分别为98.2%和96.4%,纯度分别为99.7%和96.9%。
最后,用未经多巴胺涂覆改性的原聚丙烯膜对igg/hsa进行分离,发现其并不能有效吸附蛋白,同时也没有出现蛋白洗脱,说明原聚丙烯膜不具有选择分离蛋白的能力。通过比较膜色谱介质和原聚丙烯膜,发现经多巴胺沉积制备的膜色谱介质的机械强度是原膜的2.3倍,表面接触角较原膜减小30°,同时操作压力(系统压力)较原膜减小0.2mpa(减轻了蛋白在膜面的污染)。
实施例2
在本实施例中,具备耐盐型阴离子交换膜色谱介质的制备方法如下(其制备工艺流程可以形象地以图1所示示意图来说明),具体包括以下步骤:
将0.45mg多巴胺分子加入45ml30mm碳酸钠-碳酸氢钠缓冲液中(ph10.5),同时将一片孔径为1.0微米聚醚砜膜(图1中标识1)浸入多巴胺溶液中,接着在摇床中以400转/分钟的转速在常温下反应1小时。反应结束后,取出膜片,用去离子水冲洗三遍。然后,将经多巴胺涂覆的膜片(图1中标识2)浸入2.0mg/ml三(2-氨乙基)胺和40.0mg/ml聚丙烯胺(图1中标识3)的碳酸钠-碳酸氢钠缓冲溶液(ph=11.0,40ml),在摇床中以400转/分钟的转速反应1小时(15℃)。反应结束后,用去离子水冲洗膜片。最后用氮气吹干,保存。
应用上述制备的膜色谱介质捕获和富集蛋白料液中的α1-抗胰蛋白酶(图2中标识5)(杂质蛋白为转铁蛋白、人血清白蛋白和抗凝血酶iii等(图2中标识6)),其分离过程可以图2来说明,具体分离方法和结果如下:
首先,将新制备的膜色谱介质裁剪成直径为2.5cm的膜片,将16片膜堆叠放入容器中(图2中标识4),使容器与蛋白层析系统相连接。待分离蛋白料液包含0.5mgα1-抗胰蛋白酶,总蛋白浓度为5mg/ml。蛋白溶液首先通过超滤换液,使得蛋白溶解在加有0.15m氯化钠的tris-hcl缓冲液(ph7.0,20mm)中。上述料液进样至蛋白层析系统,料液流过膜色谱介质,接着用加有0.15m氯化钠的tris-hcl缓冲液(ph7.0,20mm)淋洗膜片,最后,与膜结合的蛋白用加有1m氯化钠的tris-hcl缓冲液(ph7.0,20mm)进行线性梯度洗脱(图5为层析图谱)。收集淋洗流穿和洗脱液,进行蛋白回收率和纯度的分析(图6为电泳图谱)。
纯化结果为该耐压型膜色谱介质能够在0.15m氯化钠的高盐体系下有效分离和富集目标蛋白,获得α1-抗胰蛋白酶的回收率98.2%,纯度从原料液中的0.1mg(α1-抗胰蛋白酶)/mg(总蛋白)提高到0.6mg(α1-抗胰蛋白酶)/mg(总蛋白)。
最后,用未经多巴胺涂覆改性的原聚醚砜膜分离α1-抗胰蛋白酶,发现该膜不具有选择分离α1-抗胰蛋白酶的能力。通过比较膜色谱介质和原聚醚砜膜,发现经多巴胺沉积制备的膜色谱介质的机械强度是原膜的1.9倍,表面接触角较原膜减小32°,同时操作压力(系统压力)较原膜减小0.13mpa。
实施例3
在本实施例中,耐盐型阴离子交换膜色谱介质的制备方法(其制备工艺流程可以形象地以图1所示示意图来说明)具体包括以下步骤:
将90mg多巴胺分子加入50ml30mmtris-hcl缓冲液中(ph7.2),同时将一片孔径为5.0微米聚偏二氟乙烯膜(图1中标识1)浸入多巴胺溶液中,接着在摇床中以20转/分钟的转速在常温下反应36小时。反应结束后,取出膜片,用去离子水冲洗三遍。然后,将经多巴胺涂覆的膜片(图1中标识2)浸入2.5mg/ml胍基丁胺和0.1mg/ml聚亚己基双胍(图1中标识3)的tris-hcl缓冲溶液(ph=7.0,50ml),在摇床中以20转/分钟的转速反应48小时(80℃)。反应结束后,用去离子水冲洗膜片。最后用氮气吹干,保存。
应用上述制备的膜色谱介质对发酵料液中的重组流感疫苗(图2中标识5)进行分离纯化,其分离过程可以图2来说明,具体分离方法和结果如下:
首先,将新制备的膜色谱介质裁剪成直径为2.5cm的膜片,将八片膜堆叠(图2中标识4)放入容器中,使容器与蛋白层析系统相连接。待分离发酵料液(10mmph=7.4tris-hcl缓冲溶液,100-200mmnacl)经0.45微滤膜过滤,除去大颗粒杂质。将上述料液上样至蛋白层析系统,料液流过膜色谱介质,接着用加有150mm氯化钠的缓冲液(10mmph=7.4tris-hcl缓冲溶液)淋洗膜片,最后,与膜结合的蛋白用加有1.2m氯化钠的缓冲液(10mmph=7.4tris-hcl缓冲溶液)进行线性梯度洗脱(图7为层析图谱)。收集洗脱液,疫苗回收率和杂质总蛋白(图2中标识6)进行分析。
纯化结果为该耐盐型阴离子交换膜色谱介质能够在100-200mm氯化钠的高盐体系下对重组流感疫苗进行有效分离和富集,获得流感疫苗的活性回收率为85.4%,杂质总蛋白去除率为70.2%。
用原聚偏二氟乙烯膜对重组流感疫苗进行分离,发现其并不能结合目标分子,因此该膜不具有选择分离重组流感疫苗的能力。通过比较膜色谱介质和原聚偏二氟乙烯膜,发现经多巴胺沉积制备的膜色谱介质的机械强度是原膜的2.4倍,表面接触角较原膜减小36°,同时操作压力(系统压力)较原膜减小0.26mpa。
申请人声明,本发明通过上述实施例来说明本发明的详细工艺设备和工艺流程,但本发明并不局限于上述详细工艺设备和工艺流程,即不意味着本发明必须依赖上述详细工艺设备和工艺流程才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!