一种CO加氢制低碳混合醇的催化剂及其制备方法与流程
本发明涉及一种催化转化合成气制液体燃料或化工产品的技术,属能源化工领域。特别涉及合成气催化转化制低碳混合醇及液体烃类用催化剂及其制备方法。更具体地,涉及一种以碳为载体负载的co&co2c催化剂,活性组分为co2c和金属co的纳米复合颗粒,同时sio2为助剂组分。
背景技术:
低碳混合醇(简称低碳醇)是指由c1-c6直链伯醇组成的液体混合物,可用作替代燃料,洁净汽油添加剂或者化学品及化工原料。由合成气直接合成低碳醇的研究始于19世纪20年代,上世纪70年代的两次石油危机的影响和人们环保意识的增强,使以煤,天然气或生物质为原料经合成气制取低碳醇的研究课题受到了人们极大地重视。因方法具有较高的经济效益和资源利用优势,自上世纪初以来,研究者开发出了多种不同的合成低碳醇催化剂体系,主要包括:(1)改性高温(高压)甲醇合成催化剂。由zno/cr2o3加入碱性助剂(如cs,k)改性而得。该类催化剂主要产物为甲醇和支链异丁醇,反应条件较为苛刻(压力14-20mpa,温度350-450℃)。(2)改性低温(低压)甲醇合成催化剂。在低温合成甲醇铜基催化剂(cu/zno/al2o3或cu/zno/cr2o3)中加入碱性助剂如(如cs,k)改性同样可以提高反应产物中低碳醇的选择性。该类催化剂反应条件温和,但活性组分cu易烧结,而且产物中甲醇含量高。以上两类参见相关专利ep-0034338-a2,uspatent4513100等。(3)cu-co催化剂。法国石油研究院(ifp)首先开发了cu-co共沉淀低碳醇催化剂,目前已获得了多个专利(uspatent4122110,4291126),该类催化剂操作条件比较温和,主要产物为直链正构醇,反应活性和c2+醇的选择性较高,但总醇选择性较低且产物中水较多。(4)钼基催化剂。典型的钼基催化剂可见uspatent4882360。钼基催化剂一般具有独特的抗硫性能且不易积碳,产物含水量少,可加入碱金属助剂以及其他金属组分进行优化,可使醇的c2+/c1比例较为合理,但此催化体系对合成气比例要求苛刻,而且助剂钴极易与一氧化碳形成羰基钴造成钴流失。(5)co2c与金属co复合体系。中国专利cn105582970a报道了一种sio2或al2o3负载的co2c与co催化剂,虽然该催化剂体系甲醇在总醇中的分布较上述4种体系有所降低,但是该催化剂制备步骤冗长,而且有碳链长度大于6个碳数的醇生成,导致了醇的分离困难。sio2被研究者常以载体形式应用于费托反应中,因其具有一定的比表面积和较大的孔径,其孔道以中孔为主,并且可以在2-50nm中孔范围内进行一定程度的调节和控制,可以满足催化反应的要求。sio2载体也被用来担载钴基催化剂,用于合成气转化领域,但是其co加氢的产物一般为烃类,很少含有醇类含氧化合物。
以上合成气为原料制低碳醇的专利技术中,总醇的时空收率普遍在0.2-0.6g醇/ml催化剂/h之间,所以现有技术表明目前仍然需要研制新的合成醇的催化剂体系,改善和提高co加氢合成醇的性能,尤其是在降低甲醇的比重的同时控制醇的分布同时提高混合醇的时空收率。
金属有机框架材料(metal-organicframeworks,简称mof)是一种由金属离子与有机配体通过配位相连形成的新型多孔网状结构材料。它是利用有机配体与金属离子或金属团簇通过配位键或分子间相互作用力自组装而成的具有一维、二维或三维的无限网络结构的晶体材料。因其具有很大的比表面积、可调的孔径大小以及可根据目标要求作化学修饰、结构丰富等优点,mof在co吸附分离方面表现出很大的应用潜力。
近年来,将金属有机骨架热解制备成碳载金属或金属氧化物催化剂成为催化领域研究的热点之一。由含钴的金属有机骨架热解制备的碳载钴催化剂具有以下特点:(1)在惰性气氛条件下热解后,有机配体形成碳载体,而钴物种在碳载体上形成尺寸均一的钴纳米颗粒,而且由于碳的阻隔作用钴纳米颗粒不会发生显著迁移与团聚;(2)该类材料上钴的质量含量较高,可达40%左右;(3)惰性气氛中热解所得材料的体积急剧缩小,且继承了母体质量轻的特点,从而使所得材料的单位质量或体积上纳米钴颗粒数量较多,即具有很高的比活性位密度。现有技术文献中(nanoscale,2012,4,591-599;acscatalysis,2015,5,884-891;carmcomm,2015,51,2331-2334;化学进展,2015,27,174-191.)有大量此类材料的报道。(4)专利cn106140165a报道了以多孔碳负载双晶相钴基费托合成催化剂,研究表明钴金属活性相包含fcc与hcp两种晶相,应用于费托合成反应时,具有较高的活性和良好的c5+选择性,特别是具有极高的c5+时空收率,其中c5+产物主要以汽油和柴油组分为主。显然该专利报道的催化剂起到促进长链烃增长,高效获得c5+产物,对汽油和柴油工业应用起到一定的借鉴意义,但对于如何抑制长链烃增长,尤其获得低碳醇方面还需进一步研究。
技术实现要素:
本发明的目的就是要提供一种用于合成气制低碳醇的负载型钴基催化剂及其制备方法,该催化剂是以co-mof-71有机金属骨架为牺牲模板制备,经有机配体获得热解碳载co中间态材料,经纯co碳化后得到碳载co2c前驱体,后由合成气活化处理获得分布均匀的co和co2c纳米复合颗粒,颗粒之间排列密集,co与co2c物种之间属于动态平衡,为催化剂的活性中心;sio2是经正硅酸乙酯热解获得,以嵌入式分布在活性组分颗粒表面。本发明中以co2c与co的纳米复合颗粒为真实活性中心,复合颗粒的尺寸均一,按本发明制备的催化剂用于co加氢制低碳醇具有较高的活性和低碳醇的选择性,较低的甲醇分布,较好的总醇时空收率。
本发明利用了热解金属有机骨架和正硅酸乙酯所得材料的特点,使催化剂的co、sio2分散性好,颗粒密集,尺寸均一,钴颗粒之间由碳阻隔而不发生团聚,进而经过碳化以及活化处理可形成co与co2c纳米复合颗粒活性位,使得本发明制得的催化剂用于合成气转化制低碳混合醇反应时,催化活性高,低碳醇选择性较好,特别是总醇的时空收率较高。
为了实现本发明的上述目的,本发明提供了如下技术方案:一种co加氢制低碳混合醇的催化剂由载体、活性组分和助剂组成;载体为碳,活性组分为金属co和碳化钴co2c的纳米复合颗粒,助剂为sio2,co的质量含量为0.4-2.1wt%,co2c的质量含量为37.7-42.0%,sio2的质量含量为0-14.3%,其余为碳。
催化剂的制备方法包括如下步骤:(1)co-mof-71有机金属骨架材料的制备;(2)以co-mof-71为牺牲模板和以正硅酸乙酯为硅源,经物理化学反应制备含有si元素的催化剂前驱体;(3)活化催化剂前驱体,得到含sio2助剂的碳载co&co2c催化剂。
制备方法步骤(1)中co-mof-71有机金属骨架材料的制备,包括如下步骤:
将硝酸钴和对苯二甲酸置于含有n,n-二甲基甲酰胺和无水乙醇混合溶液的密闭容器内,将容器加热到指定温度并保持一定时间,之后趁热将所得物料过滤,并采用n,n-二甲基甲酰胺洗涤三次,最后将所得物料干燥,获得co-mof-71有机金属骨架牺牲模板材料。
优选地,所述硝酸钴、对苯二甲酸、n,n-二甲基甲酰胺和无水乙醇的摩尔比为1:1.5:95.5:31.3-1:1.5:191.0:62.6,所述的加热到指定温度并保持一定时间是指加热到110-120℃后保持12-15h。
步骤(2)中催化剂前驱体是以co2c的形式保存并负载于碳载体上,所述碳载体源于co-mof-71有机金属骨架材料中有机配体的热解,si元素源于正硅酸乙酯。以碳载co2c为催化剂前驱体,经活化得到以co与co2c纳米复合颗粒为活性组分的碳载co&co2c催化剂。催化剂前驱体的制备方法,包括如下步骤:
将co-mof-71有机金属骨架用作牺牲模板,将其浸渍于正硅酸乙酯与无水乙醇的混合溶液中,搅拌一定时间后加入氨水,继而将所得物料在空气中干燥,然后置于固定床不锈钢反应器内,于ar气氛中升温至指定温度原位热解一定时间,然后降温至室温,切入纯co气体后在一定条件下碳化,碳化完成后然后降温至室温,然后切入o2与ar的混合气钝化,得到碳载co2c前驱体。
优选地,所述干燥是于80-100℃的空气中干燥6-12h。
优选地,所述的氨水、无水乙醇、正硅酸乙酯和co-mof-71物料的质量比例为:0-1:0.92:3.7:23.6,所述的搅拌一定时间是指1-10min。
优选地,所述ar气氛中升温至指定温度原位热解一定时间是指升温至500-600℃,升温速率为1-5℃/min。
优选地,所述的纯co气体的空速为24-30l/h/g-样品、压力位2-3mpa,碳化温度为250-280℃,升温速率为1-5℃/min,碳化时间为120-140h。
优选地,所述o2与ar混合气体中o2与ar的体积比为2:98-1:99,流量维持在5-10ml/min,所述钝化的时间为10-12h。
制备方法步骤(3),活化催化剂前驱体的方法为:将催化剂前驱体置于h2与co的体积比为1-2、压力为2.8-3mpa、温度为280-300℃、空速为20-30l/h/g-催化剂的条件下,于固定床反应器下活化,升温速率为5-10℃/min,活化时间为4-6h,得到含sio2助剂的碳载co&co2c催化剂。活化后催化剂是由co和co2c是纳米级复合颗粒组成,颗粒之间排列密集,反应时,存在部分co碳化为co2c,部分co2c还原为金属co,达到co与co2c物种之间的动态平衡,原位形成了催化剂的活性中心。
一种co加氢制低碳混合醇的催化剂应用于以合成气为原料制备低碳混合醇,该应用优选控制h2与co进料体积比为1-2,反应温度为280-320℃,反应压力为2.8-3mpa,反应总空速为20-30l/h/g-催化剂。
本发明用于合成气制低碳醇反应的碳载co&co2c催化剂,反应前不需氢气还原,而是用合成气气氛处理,然后进行反应。
与现有技术相比,本发明具有以下优点:
(1)本技术将金属有机骨架热解而来的碳载co材料转化为碳载co2c前驱体材料;
(2)与现有的负载型co2c基催化剂的制备方法(cn105582970a)相比,本发明的制备方法简单,co与co2c形成纳米复合颗粒,醇的分布窄,醇的时空收率良好;
(3)sio2助剂的引入是原位引入的,将硅源正硅酸乙酯同co-mof-71有机金属骨架材料等原料共同置于固定床反应器中,与特定温度下热解反应制得含有si元素的催化剂前驱体,不用于浸渍法获得助剂,改法提高了助剂的分散度,以嵌入的形式分布在催化剂活性组分颗粒表面,对催化活性起到较佳的协同作用。
附图说明
图1为本发明实施例1-6中催化剂前驱体以及对比例1-2中催化剂的xrd图。其中(a)为实施例1中催化剂前驱体的xrd图;(b)为实施例2中催化剂前驱体的xrd图;(c)为实施例3中催化剂前驱体的xrd图;(d)为实施例4中催化剂前驱体的xrd图;(e)为实施例5中催化剂前驱体的xrd图;(f)为实施例6中催化剂前驱体的xrd图;(g)为对比例1中催化剂的xrd图;(h)为对比例2中催化剂的xrd图。可见,实施例中各催化剂均只含有co2c的衍射峰,表明催化剂在制备后以碳载co2c的形态为前驱体。
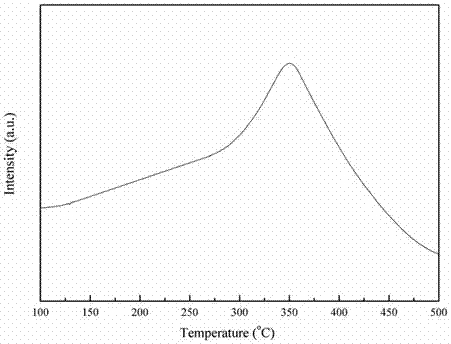
图2为本发明中以实施例1为例给出了co2c与氢气反应释放ch4气体的信号图。
图3为本发明中以实施例1与实施例2为例给出了催化剂的tem和高分辨tem图,其中图中标记a为实施例1中催化剂的tem图;b为实施例1中催化剂的高分辨tem图;c为实施例2中催化剂的tem图;d为实施例2中催化剂的高分辨tem图。
图4为本发明中以实施例1与2为例给出了催化剂上的co物种的xps表征结果。图中(1)为实施例1中催化剂表面co的2p轨道特征xps谱峰;(2)为实施例2中催化剂表面co的2p轨道特征xps谱峰。
图5为本发明实施例3-6中催化剂上硅物种的2p轨道的xps谱峰。其中,图中(1)为实施例3中催化剂表面以二氧化硅形式存在的si的2p轨道特征xps谱峰;(2)为实施例4中催化剂表面以二氧化硅形式存在的si的2p轨道特征xps谱峰;(3)为实施例5中催化剂表面以二氧化硅形式存在的si的2p轨道特征xps谱峰;(4)为实施例6中催化剂表面以二氧化硅形式存在的si的2p轨道特征xps谱峰。
具体实施方式
为更好地理解本发明,下面结合实施例和附图对本发明作进一步的说明,但本发明实施方式不限如此。
实施例1
将0.8g六水合硝酸钴(co(no3)2·6h2o)和0.46g对苯二甲酸(h2bdc)置于150ml史莱克管内,加入20mln,n-二甲基甲酰胺和5ml无水乙醇形成的混合溶液,密封后升温至110℃保持15h,然后趁热过滤,采用100mln,n-二甲基甲酰胺洗涤三次,最后于空气中100℃干燥6h,得到co-mof-71金属有机骨架物料。
将1.48gco-mof-71置于固定床不锈钢反应器内,于ar气氛中以5℃/min的升温速率升温至500℃原位热解8h,得到碳载co中间体材料。此材料中co元素的质量百分含量由原子吸收光谱(aas)表征测定为27.0%。热解完成后降温至室温,切入空速为24l/h/g-样品的纯co气体,升压至2mpa,以1℃/min的升温速率升温至250℃,保持120h,然后降温至室温,采用流量为10ml/min的1%o2/ar混合气钝化10h,得到碳载co2c前驱体。
将0.1g碳载co2c前驱体材料置于压力为3mpa,空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中,以5℃/min的升温速度升温至300℃,然后保持4h进行预处理,得到碳载co&co2c催化剂,该催化剂的co2c质量百分含量由h2的程序升温表征测定为41.7%,依据是根据公式co2c+h2–co+ch4通过测定ch4的峰面积,依据co元素质量守恒可得此催化剂中金属co的质量百分含量为1.8%,然后与基准的ch4峰面积进行校订而得,此催化剂在压力为3mpa、温度为300℃、空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中进行费托合成反应,结果见下表1。
该催化剂前驱体的晶相结构由xrd表征后示于图1,从图1中可以看到只有co2c形态的特征峰出现,预处理后的催化剂的tem图见图2,可见众多以纳米颗粒形态出现的活性位,将其中某一颗粒采用高分辨电镜放大后示于图3,可见单一颗中同时含有co2c和co,预处理后的催化剂的xps表征结果见图4,从图4中可见金属co和co2c物种中co的特征峰出现,以上说明催化剂的活性位以co2c的形式为前驱体,而真实活性位为co与co2c纳米复合颗粒。图2可知测定催化剂上co2c物种的含量。
实施例2
将0.8g六水合硝酸钴(co(no3)2·6h2o)和0.46g对苯二甲酸(h2bdc)置于150ml史莱克管内,加入40mln,n-二甲基甲酰胺和10ml无水乙醇形成的混合溶液,密封后升温至120℃保持12h,然后趁热过滤,采用100mln,n-二甲基甲酰胺洗涤三次,最后于空气中80℃干燥12h,得到co-mof-71金属有机骨架物料。
将1.48gco-mof-71置于固定床不锈钢反应器内,于ar气氛中以1℃/min的升温速率升温至600℃原位热解4h,得到碳载co中间体材料,此材料中co元素的质量百分含量由aas表征测定为33.1%,热解完成后降温至室温,切入空速为30l/h/g-样品的纯co气体,升压至3mpa,以5℃/min的升温速率升温至280℃,保持140h,然后降温至室温,采用流量为5ml/min的2%o2/ar混合气钝化12h,得到碳载co2c前驱体。
将0.1g碳载co2c前驱体材料置于压力为2.8mpa,空速为20l/h/g-催化剂的合成气(h2与co体积比为1)中,以5℃/min的升温速度升温至280℃,然后保持6h进行预处理,得到碳载co&co2c催化剂,该催化剂的co2c质量百分含量由h2的程序升温表征测定为51.4%,依据是根据公式co2c+h2–co+ch4通过测定ch4的峰面积,然后与基准的ch4峰面积进行校订而得,依据co元素质量守恒可得此催化剂中金属co的质量百分含量为0.4%,此催化剂在压力为2.8mpa、温度为280℃、空速为20l/h/g-催化剂的合成气(h2与co体积比为1)中进行费托合成反应,结果见下表1。
催化剂前驱体的晶相结构由xrd表征后示于图1,从图1中可以看到只有co2c形态的特征峰出现,预处理后的催化剂的tem图见图2,可见众多以纳米颗粒形态出现的活性位,将其中某一颗粒采用高分辨电镜放大后示于图3,可见单一颗中同时含有co2c和co,预处理后的催化剂的xps表征结果见图4,从图4中可见金属co和co2c物种中co的特征峰出现,以上说明催化剂的活性位以co2c的形式为前驱体,而真实活性位为co与co2c纳米复合颗粒。图3可知,催化剂上存在众多纳米颗粒活性位,同时各纳米颗粒活性位由co与co2c物种复合而成。图4可知,,催化剂上co物种同时以co与co2c存在。
实施例3
将0.8g六水合硝酸钴(co(no3)2·6h2o)和0.46g对苯二甲酸(h2bdc)置于150ml史莱克管内,加入30mln,n-二甲基甲酰胺和10ml无水乙醇形成的混合溶液,密封后升温至110℃保持15h,然后趁热过滤,采用100mln,n-二甲基甲酰胺洗涤三次,最后于空气中80℃干燥12h,得到co-mof-71金属有机骨架物料。
按照下述步骤制备含二氧化硅助剂质量含量为14.1%的碳载co&co2c催化剂:称取0.070g正硅酸乙酯置于1.58g无水乙醇中形成混合溶液,加入0.37gco-mof-71,室温搅拌10min,然后加入0.1g氨水,之后在空气中于80℃干燥12h,随之将所得物料置于固定床反应器内,在ar下以5℃/min升温至500℃并保持8h,此材料中co元素的质量百分含量采用aas表征测试为23.2%,热解完成后降温至室温,切入空速为30l/h/g-样品的纯co气体,升压至2mpa,以5℃/min的升温速率升温至250℃,保持120h,然后降温至室温,采用流量为10ml/min的1%o2/ar混合气钝化10h,得到含二氧化硅助剂的碳载co2c前驱体;将0.1g此前驱体材料置于压力为3mpa,空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中,以5℃/min的升温速度升温至300℃,然后保持4h进行预处理,得到含二氧化硅助剂的碳载co&co2c催化剂,该催化剂的co2c质量百分含量由h2的程序升温表征测定为37.7%,依据是根据公式co2c+h2–co+ch4通过测定ch4的峰面积,然后与基准的ch4峰面积进行校订而得,依据co元素质量守恒可得此催化剂中金属co的质量百分含量为1.2%,根据正硅酸乙酯添加量测算二氧化硅助剂的质量百分含量为11.2%,此催化剂在压力为3mpa、温度为300℃、空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中进行费托合成反应,结果见下表1。
催化剂前驱体的晶相结构由xrd表征后示于图1,从图1中可以看到只有co2c形态的特征峰出现,而真实催化剂的活性位与实施例1、2具有相同的特点(此处省略表征结果),催化剂上二氧化硅物种的xps表征结果如图5所示,可见si的2p轨道结合能为102ev的特征峰,可定性为二氧化硅物种,说明催化剂表面上存在二氧化硅助剂。
实施例4
将0.8g六水合硝酸钴(co(no3)2·6h2o)和0.46g对苯二甲酸(h2bdc)置于150ml史莱克管内,加入20mln,n-二甲基甲酰胺和5ml无水乙醇形成的混合溶液,密封后升温至110℃保持15h,然后趁热过滤,采用100mln,n-二甲基甲酰胺洗涤三次,最后于空气中90℃干燥10h,得到co-mof-71金属有机骨架物料。
按照下述步骤制备含二氧化硅助剂质量百分含量为12.9%的co&co2c催化剂:称取0.070g正硅酸乙酯置于1.58g无水乙醇中形成混合溶液,加入0.37gco-mof-71,室温搅拌5min,然后加入0.1g氨水,之后在空气中于80℃干燥12h,随之将所得物料置于固定床反应器内,在ar下以5℃/min升温至600℃并保持8h,此材料中co元素的质量百分含量采用aas表征测试为27.5%,热解完成后降温至室温,切入空速为30l/h/g-样品的纯co气体,升压至2mpa,以5℃/min的升温速率升温至250℃,保持120h,然后降温至室温,采用流量为10ml/min的1%o2/ar混合气钝化10h,得到含二氧化硅助剂的碳载co2c前驱体;将0.1g此前驱体材料置于压力为3mpa,空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中,以5℃/min的升温速度升温至300℃,然后保持4h进行预处理,得到含二氧化硅助剂的碳载co&co2c催化剂,该催化剂的co2c质量百分含量由h2的程序升温表征测定为42.0%,依据是根据公式co2c+h2–co+ch4通过测定ch4的峰面积,然后与基准的ch4峰面积进行校订而得,依据co元素质量守恒可得此催化剂中金属co的质量百分含量为2.1%,根据正硅酸乙酯添加量测算二氧化硅助剂的质量百分含量为12.9%,此催化剂在压力为3mpa、温度为300℃、空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中进行费托合成反应,结果见下表1。
催化剂前驱体的晶相结构由xrd表征后示于图1,从图1中可以看到只有co2c形态的特征峰出现,而真实催化剂的活性位与实施例1、2具有相同的特点(此处省略表征结果),催化剂上二氧化硅物种的xps表征结果如图5所示,可见si的2p轨道结合能为102ev的特征峰,可定性为二氧化硅物种,说明催化剂表面上存在二氧化硅助剂。
实施例5
将0.8g六水合硝酸钴(co(no3)2·6h2o)和0.46g对苯二甲酸(h2bdc)置于150ml史莱克管内,加入20mln,n-二甲基甲酰胺和5ml无水乙醇形成的混合溶液,密封后升温至110℃保持15h,然后趁热过滤,采用100mln,n-二甲基甲酰胺洗涤三次,最后于空气中100℃干燥12h,得到co-mof-71金属有机骨架物料。
按照下述步骤制备含二氧化硅助剂质量百分含量为7.7%的co&co2c催化剂:称取0.045g正硅酸乙酯置于0.79g无水乙醇中形成混合溶液,加入0.37gco-mof-71,室温搅拌5min,然后加入0.1g氨水,之后在空气中于80℃干燥12h,随之将所得物料置于固定床反应器内,在ar下以5℃/min升温至500℃并保持8h,此材料中co元素的质量百分含量采用aas表征测试为24.3%,热解完成后降温至室温,切入空速为30l/h/g-样品的纯co气体,升压至2mpa,以5℃/min的升温速率升温至250℃,保持120h,然后降温至室温,采用流量为10ml/min的1%o2/ar混合气钝化10h,得到含二氧化硅助剂的碳载co2c前驱体;将0.1g此前驱体材料置于压力为3mpa,空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中,以5℃/min的升温速度升温至300℃,然后保持4h进行预处理,得到含二氧化硅助剂的碳载co&co2c催化剂,该催化剂的co2c质量百分含量由h2的程序升温表征测定为39.5%,依据是根据公式co2c+h2–co+ch4通过测定ch4的峰面积,然后与基准的ch4峰面积进行校订而得,依据co元素质量守恒可得此催化剂中金属co的质量百分含量为1.1%,根据正硅酸乙酯添加量测算二氧化硅助剂的质量百分含量为7.7%,此催化剂在压力为3mpa、温度为300℃、空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中进行费托合成反应,结果见下表1。
催化剂前驱体的晶相结构由xrd表征后示于图1,从图1中可以看到只有co2c形态的特征峰出现,而真实催化剂的活性位与实施例1、2具有相同的特点(此处省略表征结果),催化剂上二氧化硅物种的xps表征结果如图5所示,可见si的2p轨道结合能为102ev的特征峰,可定性为二氧化硅物种,说明催化剂表面上存在二氧化硅助剂。
实施例6
将0.8g六水合硝酸钴(co(no3)2·6h2o)和0.46g对苯二甲酸(h2bdc)置于150ml史莱克管内,加入40mln,n-二甲基甲酰胺和10ml无水乙醇形成的混合溶液,密封后升温至110℃保持15h,然后趁热过滤,采用100mln,n-二甲基甲酰胺洗涤三次,最后于空气中100℃干燥12h,得到co-mof-71金属有机骨架物料。
按照下述步骤制备含二氧化硅助剂质量百分含量为14.3%的co&co2c催化剂:称取0.092g正硅酸乙酯置于0.79g无水乙醇中形成混合溶液,加入0.37gco-mof-71,室温搅拌10min,然后加入0.1g氨水,之后在空气中于100℃干燥12h,随之将所得物料置于固定床反应器内,在ar下以5℃/min升温至500℃并保持8h,此材料中co元素的质量百分含量采用aas表征测试为22.1%,热解完成后降温至室温,切入空速为30l/h/g-样品的纯co气体,升压至2mpa,以5℃/min的升温速率升温至250℃,保持120h,然后降温至室温,采用流量为10ml/min的1%o2/ar混合气钝化10h,得到含二氧化硅助剂的碳载co2c前驱体;将0.1g此前驱体材料置于压力为3mpa,空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中,以5℃/min的升温速度升温至300℃,然后保持4h进行预处理,得到含二氧化硅助剂的碳载co&co2c催化剂,该催化剂的co2c质量百分含量由h2的程序升温表征测定为37.5%,依据是根据公式co2c+h2–co+ch4通过测定ch4的峰面积,然后与基准的ch4峰面积进行校订而得,依据co元素质量守恒可得此催化剂中金属co的质量百分含量为0.5%,根据正硅酸乙酯添加量测算二氧化硅助剂的质量百分含量为7.7%,此催化剂在压力为3mpa、温度为300℃、空速为30l/h/g-催化剂的合成气(h2与co体积比为2)中进行费托合成反应,结果见下表1。
催化剂前驱体的晶相结构由xrd表征后示于图1,从图1中可以看到只有co2c形态的特征峰出现,而真实催化剂的活性位与实施例1、2具有相同的特点(此处省略表征结果),催化剂上二氧化硅物种的xps表征结果如图5所示,可见si的2p轨道结合能为102ev的特征峰,可定性为二氧化硅物种,说明催化剂表面上存在二氧化硅助剂。
对比例1
以实施例1中的合成的co-mof-71为牺牲模板。将0.37gco-mof-71置于固定床反应器内,于ar气氛中以5℃/min的升温速率升温至500℃原位热解8h,得到碳载co中间体材料,此材料中co的质量百分含量采用aas表征测定为27.0%。热解完成后切入反应温度为300℃、h2与co体积进料比2、进气空速为30l/h/g催化剂、反应压力为3mpa的反应条件进行评价。在上述条件下,该催化剂的性能评价结果见下表1。
该催化剂晶相结构的xrd表征结果见图1,从图1中可以看到只有金属钴的特征峰出现。
对比例2
以实施例1中的合成的co-mof-71为牺牲模板。将0.37gco-mof-71置于固定床反应器内,于ar气氛中以1℃/min的升温速率升温至600℃原位热解8h,得到碳载co中间体材料,此材料中co的质量百分含量采用aas表征测试为33.1%,热解完成后切入反应温度为300℃、h2与co体积进料比2、进气空速为30l/h/g催化剂、反应压力为3mpa的反应条件进行评价。在上述条件下,该催化剂的性能评价结果见下表1。
该催化剂晶相结构的xrd表征结果见图1,从图1中可以看到只有金属钴的特征峰出现。
表1实施例1-6和对比例1-2的催化剂上合成气制低碳醇反应结果
由表1可见,实施例中采用碳载的co&co2c催化剂表现出了良好的合成气制合成醇的反应性能。其中,甲醇的选择性低于15%,c2-c6低碳醇的选择性大于26.7%,特别是醇的时空收率大于0.20g/h/g-催化剂。对比例1与对比2提供的催化剂为碳负载的钴基催化剂,有机产品全部为烃类化合物,说明本发明提供的催化剂主要源于本发明独特的制备方法。
本发明中真实的催化剂上同时包含co和co2c两种,助剂sio2分散均匀。另外,本发明中催化剂的前驱体以多孔炭载碳化钴的形式存在,从而避免了催化剂暴露于空气中被氧化失活。从表1中催化剂的催化性能可以看出,本发明方法提供的多孔炭载钴催化剂,与由相同的条件热解而来的多孔炭载钴催化剂相比,在相同的反应条件下,副产物甲烷以及co2的选择性得到了明显降低,而催化剂的活性,c5+时产物很少,链增长得到有效抑制,低碳醇收率显著增加。
- 还没有人留言评论。精彩留言会获得点赞!