一种催化苄基化反应新方法与流程
本发明涉及一种催化苄基化反应新方法。具体来说是采用具有强
背景技术:
通过friedel-crafts烷基化反应构建c-c单键是工业有机合成重要的单元反应之一。其中芳烃经friedel-crafts苄基化制备的二芳基甲烷及其衍生物是十分具有商业意义的精细化学品,在医药、染料、香料等行业中有广泛应用(bandinim,mellonia,umani-ronchia.angewandtechemieinternationaledition,2004,43(5):550-556.;rothbj,manolaj,dreicerr,etal.investigationalnewdrugs,2002,20(4):425-429.)。工业生产中往往使用产生卤化氢气体的苄基卤作为苄基化试剂,并使用alcl3,fecl3和h2so4等无机均相酸催化剂,存在催化剂一次消耗,污染、腐蚀严重等问题(kumarcr,raoktv,prasadpss,etal.journalofmolecularcatalysisa:chemical,2011,337(1):17-24.)。苄醇是替代苄基卤的理想清洁苄基化试剂,但其活性较弱,在催化剂酸催化能力不足时难以发生与芳烃的苄基化反应,或易发生自身脱水生成二苄醚(deshpandeab,bajpaiar,samantsd.appliedcatalysisa:general,2001,209(1):229-235.)。
沸石分子筛、固体超强酸、负载型催化剂等固体酸催化剂虽然对苄基化反应有着较好的催化活性,并且易于分离和回收,但存在制备过程复杂,制备重现性差,易失活、传质和扩散阻力大等问题,且在苄醇路线苄基化反应的过程中,副产物水容易导致催化活性组分的流失(liq,jiangs,jis,etal.journalofporousmaterials,2015,22(5):1205-1214.)。酸功能化离子液体虽兼具均相与多相催化的优势,但其由磺酸基等官能团引入的
综上所述,采用苄醇路线苄基化反应方法制备二苯基甲烷及其衍生物的研究具有重要意义,开发新的环境友好催化方法,实现芳环化合物与苄醇的高效催化,及催化剂的简易分离和稳定重复使用,是精细有机合成领域的现实需求。
技术实现要素:
为解决苄醇路线的苯环化合物苄基化反应活性低,催化剂耐水性和重复使用性差等问题,本发明提出了一种采用强
本发明的技术方案为:
按物质的量比n(苯环化合物):n(苄醇):n(催化剂)=15:1:2.3×10-2的比例,称取苯环化合物反应底物、苄醇和催化剂加入反应瓶中,此时反应体系为均相。在回流温度下搅拌反应1h后冷却,体系变为有机相和水相两相。分别回收上层有机相和下层水相,水相中的催化剂经烘干除水后,即可用于循环使用。
上述技术方案中的所述催化剂为具有强
按物质的量比n(三氟甲磺酸):n(氯化胆碱)=2:1的比例,在冰水浴条件下,将三氟甲磺酸加入氯化胆碱中,保温10min后,在油浴中加热至80℃反应2h,得到浅黄色液体即为催化剂[chcl][tfoh]2。
本发明与现有的friedel-crafts苄基化制备的二苯基甲烷及其衍生物的技术相比,具有以下优点:
1、采用环境友好的低温共熔体催化剂催化苄醇与苯环化合物的苄基化反应,反应不产生卤化氢等污染腐蚀性副产物,原子经济性高,环境因子低。
2、低温共熔体催化剂[chcl][tfoh]2制备简单,具有其氢键供体三氟甲磺酸带来的强酸性和催化活性,但其稳定性却较三氟甲磺酸大大提高,遇水后可保持结构和催化性能的稳定。
3、低温共熔体催化剂[chcl][tfoh]2不溶于苯、甲苯、对二甲苯、邻二甲苯、间二甲苯、均三甲苯、苯甲醚等苯环化合物及其苄基化产物,而溶于苄醇和水。在反应过程中与反应体系呈均相,而在苄醇完全消耗后从有机相中析出,并溶于生成的水相,易于实现与产物的反应控制自分离和重复使用。
附图说明
图1为实施例1所制备的低温共熔体催化剂[chcl][tfoh]2及其原料的ft-ir谱图;其中(a)chcl(b)tfoh(c)[chcl][tfoh]2。
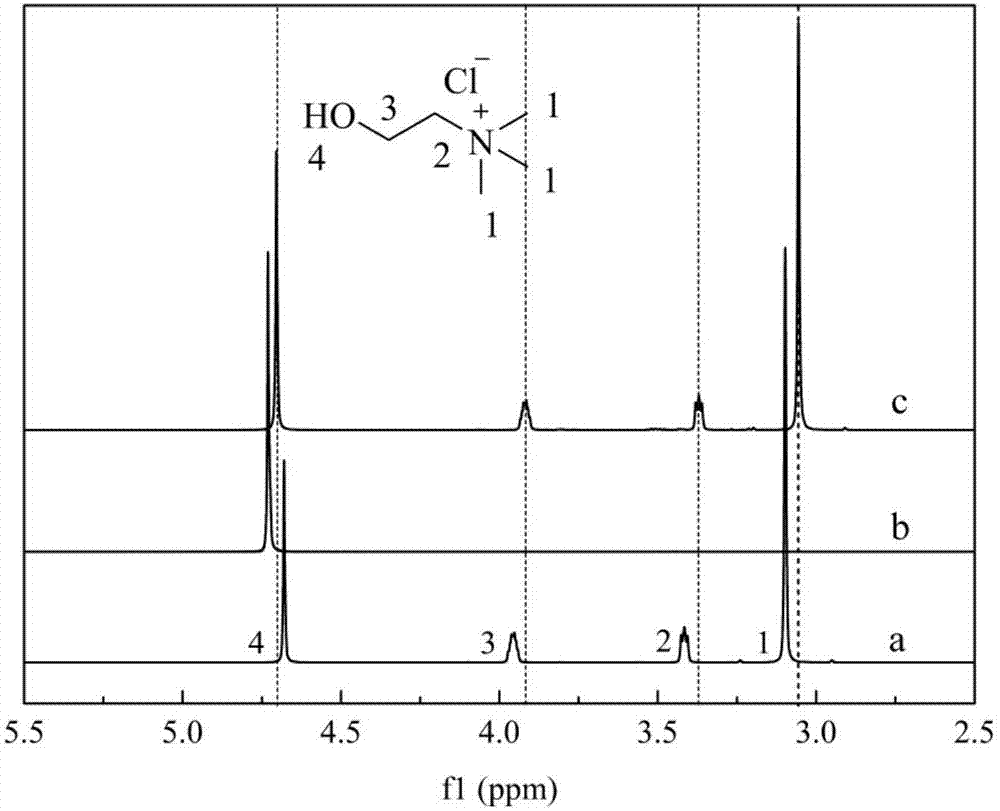
图2为实施例1所制备的低温共熔体催化剂[chcl][tfoh]2及其原料的1hnmr谱图;其中(a)chcl(b)tfoh(c)[chcl][tfoh]2。
图3为实施例1所制备的低温共熔体催化剂[chcl][tfoh]2的tg-dtg谱图。
图4为实施例2中[chcl][tfoh]2催化对二甲苯与苄醇苄基化反应前、反应后体系的宏观状态图。
具体实施方式
下面结合具体实施例进一步说明本发明,但不因此而限制本发明。
实施例1
称取氯化胆碱2.79g(0.02mol)加入50ml圆底烧瓶中,在冰水浴条件下,加入三氟甲磺酸6.00g(0.04mol)保温10min后,将圆底烧瓶置于油浴装置80℃反应2h,得到低温共熔体催化剂[chcl][tfoh]2浅黄色液体。该液体催化剂在室温至160℃范围内均不溶对二甲苯等单环芳烃和其苄基化产物,易溶于苄醇和水,相较于其原料三氟甲磺酸溶于极性溶剂中产生的强烈放热现象,并可能导致溶剂挥发甚至爆炸等问题,低温共熔体催化剂[chcl][tfoh]2稳定性大大增强,久置后仍为均一液体。
附图1所示的ft-ir(kbr,v/cm-1)谱图,[chcl][tfoh]2(c)与chcl(a)相比,-oh峰(3256cm-1到3485cm-1)和c-h峰(3018cm-1到3054cm-1)发生蓝移,并且分子间氢键峰形明显变宽,说明[chcl][tfoh]2中有氢键产生。同时,[chcl][tfoh]2(c)保持了其氢键供体tfoh(b)的所有特征峰。
附图2所示的1hnmr(d2o)谱图中,[chcl][tfoh]2(c)与chcl(a)相比1、2、3位置的氢位移值略微向高场移动,并在chcl(a)4位置中δ=4.68的峰和tfoh(b)中δ=4.73的峰变为δ=4.70的新峰。显示出[chcl][tfoh]2中chcl和tfoh之间存在氢键的相互作用。
附图3所示的tg-dtg谱图中,[chcl][tfoh]2在160℃以下结构保持稳定。
实施例2
称取实施例1中制备的低温共熔体催化剂[chcl][tfoh]20.1g,对二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h,反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表1。
附图4所示的是反应体系反应前和反应后的宏观状态图,反应时体系为均相,反应结束后,催化剂进入水相从而实现与反应产物的自分离。
实施例3
称取实施例1中制备的低温共熔体催化剂[chcl][tfoh]20.1g,邻二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h,反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表1。
实施例4
称取实施例1中制备的低温共熔体催化剂[chcl][tfoh]20.1g,间二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h,反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表1。
实施例5
称取实施例1中制备的低温共熔体催化剂[chcl][tfoh]20.1g,均三甲苯18.03g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于136℃反应1h,反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表1。
实施例6
称取实施例1中制备的低温共熔体催化剂[chcl][tfoh]20.1g,苯甲醚16.22g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于134℃反应1h,反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表1。
实施例7
称取实施例1中制备的低温共熔体催化剂[chcl][tfoh]20.1g,甲苯13.82g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于110℃反应1h,反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表1。
实施例8
称取实施例1中制备的低温共熔体催化剂[chcl][tfoh]20.1g,苯11.72g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于84℃反应1h,反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表1。
表1低温共熔体[chcl][tfoh]2催化苯环化合物的苄基化反应
实施例9-14
将实施例2中的含有催化剂的水相与上层有机相分离,置于于烘箱中100℃烘干。将此回收的低温共熔体[chcl][tfoh]2催化剂,与对二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h。反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析。循环使用实验进行6次,所得催化结果见表2。
表2低温共熔体催化剂[chcl][tfoh]2的重复使用性能
对比例1
称取1,3-丙烷磺内酯12.21g(0.10mol)加入250ml三口烧瓶中,量取100ml乙酸乙酯使其溶解,将n-甲基咪唑9.02g(0.10mol)使用恒压滴液漏斗以5d/min的速度缓慢滴加入体系,50℃机械搅拌下反应24h,反应物抽滤并用乙酸乙酯洗涤3次,随后在80℃真空干燥4h得到的白色粉末即为中间体1-甲基-3-(3-磺酸基)丙基咪唑盐(mimps)。称取磷钨酸(h3pw12o40)5.76g(0.002mol)置于100ml单口烧瓶中,量取40ml去离子水使其溶解,再加入mimps1.23g(0.006mol),磁力搅拌下室温(25℃)反应24h。减压蒸馏除去多余水分后,置于鼓风干燥箱中120℃烘干所得灰白色粉末即为对比催化剂1[mimps]3pw12o40。
称取上述对比催化剂1[mimps]3pw12o400.1g,对二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h。反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表3。
对比例2
称取1,3-丙烷磺内酯12.21g(0.10mol)加入250ml三口烧瓶中,量取100ml乙酸乙酯使其溶解,将三乙胺10.19g(0.10mol)使用恒压滴液漏斗以5d/min的速度缓慢滴加入体系,50℃机械搅拌下反应24h,反应物抽滤并用乙酸乙酯洗涤3次,随后在80℃真空干燥4h得到的白色粉末即为中间体丙基磺酸基三乙胺盐(teaps)。称取磷钨酸(h3pw12o40)5.76g(0.002mol)置于100ml单口烧瓶中,量取40ml去离子水使其溶解,再加入teaps1.34g(0.006mol),磁力搅拌下室温(25℃)反应24h。减压蒸馏除去多余水分后,置于鼓风干燥箱中120℃烘干所得灰白色粉末即为对比催化剂2[teaps]3pw12o40。
称取上述对比催化剂2[teaps]3pw12o400.1g,对二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h。反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表3。
对比例3
称取2.79g(0.02mol)氯化胆碱,7.61g(0.04mol)对甲苯磺酸一水合物加入50ml圆底烧瓶中,置于油浴装置120℃反应2h,得到无色透明液体即为对比催化剂3[chcl][p-tsoh]2。
称取上述对比催化剂3[chcl][p-tsoh]20.1g,对二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h。反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表3。
对比例4
称取2.79g(0.02mol)氯化胆碱,3.84g(0.04mol)甲磺酸加入50ml圆底烧瓶中,置于油浴装置80℃反应2h,得到无色透明液体即为对比催化剂4[chcl][ch3so3h]2。
称取上述对比催化剂4[chcl][ch3so3h]20.1g,对二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h。反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表3。
对比例5
称取2.79g(0.02mol)氯化胆碱,5.45g(0.04mol)zncl2加入50ml圆底烧瓶中,置于油浴装置120℃反应2h,得到无色透明液体即为对比催化剂5[chcl][zncl2]2。
称取上述对比催化剂5[chcl][zncl2]20.1g,对二甲苯15.93g(0.15mol),苄醇1.08g(0.01mol)置于100ml三口烧瓶中,在配有回流装置和温度计的条件下于140℃反应1h。反应结束后静置,取上层有机相加入无水硫酸镁除水后进行气相色谱分析,催化结果见表3。
表3对比催化剂催化对二甲苯与苄醇苄基化反应结果
- 还没有人留言评论。精彩留言会获得点赞!