一种降解抗生素废水的新型Z型声催化剂及其制备方法和应用与流程
本发明属于声催化领域,尤其涉及具有z型结构的声催化剂er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的合成及其在声催化降解抗生素废水中的应用。
背景技术:
近年来,人类对抗生素的滥用产生了大量含抗生素的废水,该类废水中残留的抗生素在生态环境中长期积累,不但具有致畸、致癌性,还会导致其中的病原微生物抗药性增强,对生态平衡和人类健康造成严重危害。因此,对抗生素废水的治理迫在眉睫。废水中残留的抗生素,生物毒性大,ph波动大,色度深,气味重,给废水处理带来极大的困难。面对难以被充分降解的抗生素,如何将它们彻底的无害化处理,一直是困扰科学家的难题。传统的对抗生素废水的处理采用生物处理方法、物理处理方法和化学处理方法等。但是由于一般抗生素废水浓度较高,这些方法的去除效率并不高,而且处理不彻底,容易导致二次污染。而且抗生素具有抑菌效果,采用常规的生物处理等技术难以获得理想效果。因此,找到一个新的处理方法是相当必要的。声催化由于其操作简单、彻底等优点被广泛应用于污染物的治理等领域。
声催化作为一种高级氧化过程,是利用超声在溶液中的空化效应,发生声致发光和“热点”。一方面,超声空化效应产生“热点”的瞬间,温度可以达到5000k和1000标准大气压的高压,这就可以使溶液中的水分解产生有很强氧化能力的氢氧自由基。另一方面,声致发光能够产生很宽范围的光,这些光可以激发声催化剂产生光生电子和空穴对来降解抗生素。理想的声催化降解反应所需要的催化剂必须具备稳定性、耐腐蚀、光响应范围广和带宽合适等特点。在众多声催化剂中niga2o4以其耐腐蚀性、无毒性和价格低廉的特点成为最有吸引力的声催化剂之一。但对于niga2o4作为声催化剂本身也有其不可克服的缺点和不足。由于它的禁带宽度(eg=3.54ev)较宽,因此只能吸收紫外光(λ<387nm)而被激发。遗憾的是在声致发光中,紫外光的成分相当低。bivo4是继niga2o4之后的另一个卓越的半导体催化剂,因其具有无毒、稳定、禁带宽度较小等优点被用于催化降解有机污染物。但是单纯的niga2o4和bivo4声催化降解效率也很低,是由于它们的光生电子和空穴对容易复合。因此,发明一种可以彻底抑制光生电子和空穴对的催化剂变得尤为重要。
技术实现要素:
为了解决宽带半导体niga2o4不能有效利用声致发光中的紫外光问题,本发明提供了一种将上转换发光材料er3+:y3al5o12与半导体催化剂相结合的方式。为了解决电子和空穴的复合问题,本发明设计合成一种将fe3+掺杂到niga2o4和bivo4中形成一种高效的氧化还原循环复合中心(fe3+-niga2o4||bivo4),同时负载贵金属au。这种新型z型结构的声催化剂可以大幅度提高声催化降解的效率。
本发明采用的技术方案是:一种降解抗生素废水的新型z型声催化剂,所述的降解抗生素废水的新型z型声催化剂是:er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4。优选的,ni(fe0.05ga0.95)2o4和bivo4摩尔比为1:1。
一种降解抗生素废水的新型z型声催化剂的制备方法,包括如下步骤:
1)将适量的er2o3、y2o3粉末溶解在浓硝酸中,并磁力加热搅拌至无色透明,得稀土离子溶液;按比例称取al(no3)3·9h2o溶解在蒸馏水中,在室温下用玻璃棒搅拌并慢慢加入到稀土离子溶液中,加入柠檬酸作为螯合剂和助溶剂,按摩尔比,柠檬酸:稀土离子=3:1,于50-60℃加热搅拌,当溶液呈粘稠状时停止,得发泡黏胶状溶液,将发泡黏胶状溶液放入烘箱恒温80℃加热36h,得泡沫溶胶,将泡沫溶胶在500℃加热50min,然后在1100℃煅烧2h,冷却至室温,得er3+:y3al5o12粉末。
2)将ga2o3固体加入到硝酸镍溶液中,再加入适量fe(no3)3·9h2o,产生的混合液用氢氧化钠调节ph到12,然后加入er3+:y3al5o12继续搅拌,得到的悬浮溶液转移到反应釜中,于180℃下反应48h,冷却至室温,得到的反应物用去离子水清洗,然后在80℃下烘干8h,研磨,再于500℃的马弗炉中,焙烧2h,取出,再研磨,得到er3+:y3al5o12@ni(fe0.05ga0.95)2o4纳米粉末。
3)将纳米粉末er3+:y3al5o12@ni(fe0.05ga0.95)2o4和haucl4溶液溶解在乙醇中,超声分散30分钟,得悬浮液,将悬浮液加热到60-65℃,保持恒温30分钟,过滤,洗涤,分离出来的粉末在350℃下煅烧2h,研磨,得到er3+:y3al5o12@ni(fe0.05ga0.95)2o4/au。
4)将bi(no3)3·5h2o完全溶解在硝酸中形成溶液a,nh4vo3溶解在氢氧化钠中形成溶液b,将溶液b逐滴加入到溶液a中形成悬浮溶液,充分搅拌后,调节ph=7,继续搅拌30min,然后转移到反应釜中,180℃,反应24h后,得产物,水洗,干燥,研磨,得到bivo4纳米粒子。
5)将er3+:y3al5o12@ni(fe0.05ga0.95)2o4/au和bivo4纳米粉末加入到无水乙醇中,超声分散30min,得悬浮液,将悬浮液加热到60-65℃,保持恒温30分钟,过滤,洗涤,分离出来的粉末在500℃下煅烧2h,研磨,得er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4纳米粉末。
上述的一种降解抗生素废水的新型z型声催化剂在声催化降解抗生素废水中的应用。方法为:将降解抗生素废水的新型z型声催化剂加入到含有抗生素的废水中,在室温,103650pa,50w、40khz的超声波照射,照射时间为300min。优选的,所述的抗生素为磺胺。
本发明,降解抗生素废水的新型z型声催化剂er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4在超声波照射下声催化降解磺胺的过程分析:众所周知,niga2o4的带宽为3.5ev,当声致发光产生的可见光直接照射其表面时,不能激发niga2o4粒子产生电子-空穴对,而当可见光照射到er3+:y3al5o12后,由于er3+:y3al5o12在可见光(低能量的光)照射下基态光子能够被逐级激发到更高的能级,然后这些光子再跃迁回基态后发射出紫外光(高能量的光),这些紫外光能有效地激发er3+:y3al5o12周围的niga2o4粒子生成光生电子-空穴对。为了最大限度的给niga2o4提供紫外光,使其声催化效率得到大幅度的提高,本发明利用宽波段光谱吸收上转换紫外发光材料er3+:y3al5o12,最大限度地把声致发光中产生的红外光和可见光转变成紫外光。由于niga2o4和bivo4被光激发后产生的电子-空穴对容易复合从而失去催化活性,因此为了彻底分离光生e-和h+进一步提高催化活性,本发明将fe3+掺到niga2o4中与bivo4中的v5+形成了一种假定的复合循环中心。在这个循环体系中v5+被bivo4导带上的e-还原成v4+,fe3+被niga2o4价带上的h+氧化成fe4+。此外,fe4+足以在bivo4导带和niga2o4价带形成的假定复合循环中心氧化v4+,从而再次生成fe3+和v5+重复之前的氧化还原反应。值得注意的是,niga2o4导带上的e-和bivo4价带上的h+被彻底的分开。一方面,bivo4价带生成的空穴在表面能够直接分解磺胺。另一方面niga2o4导带上的产生的e-与溶解在水溶液中氧气反应,生成超氧自由基,经过一系列的化学反应,生成氢氧自由基,氢氧自由基具有很强的氧化性,能够将磺胺氧化。本发明利用设计的假定复合循环氧化还原体系,有效的避免了光生电子和空穴的复合,从而提高催化剂的降解效率。
本发明的有益效果:
本发明制备的er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4纳米级声催化剂性质稳定,耐高温,耐酸碱腐蚀,与单纯的niga2o4和bivo4相比,本发明的催化剂在超声照射下降解抗生素的效率有了大幅度提高。相比于传统的ta2o5-tio2光催化剂,本发明的er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4纳米级声催化剂不仅具有传统声催化降解的优点,而且其最值得关注的是由于有上转换发光材料er3+:y3al5o12的加入和fe3+离子的掺杂以及au作为导电通道和助催剂的使用。上转换发光材料er3+:y3al5o12的加入使er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4纳米级声催化剂不仅可以吸收紫外光,还可以将吸收的红外光和可见光转化为紫外光。fe3+离子的掺杂可以使被光激发的催化剂光生电子和空穴对得到了更彻底的分离,大幅度的提高了声催化剂声催化降解抗生素废水的效率。
附图说明
图1a是er3+:y3al5o12的x射线粉末衍射(xrd)图。
图1b是niga2o4的x射线粉末衍射(xrd)图。
图1b-1是ni(fe0.05ga0.95)2o4的x射线粉末衍射(xrd)图。
图1c是bivo4的x射线粉末衍射(xrd)图。
图1d是er3+:y3al5o12@niga2o4的x射线粉末衍射(xrd)图。
图1d-1是er3+:y3al5o12@ni(fe0.05ga0.95)2o4的x射线粉末衍射(xrd)图。
图1e是er3+:y3al5o12@ni(fe0.05ga0.95)2o4-bivo4的x射线粉末衍射(xrd)图。
图1f是er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的x射线粉末衍射(xrd)图。
图2a是er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的透射电子显微镜(tem)图(100nm)。
图2b是er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的透射电子显微镜(tem)图(20nm)。
图2c是er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的透射电子显微镜(tem)图(10nm)。
图3a是er3+:y3al5o12的红外光谱(ir)图。
图3b是niga2o4的红外光谱(ir)图。
图3c是bivo4的红外光谱(ir)图。
图3d是ni(fe0.05ga0.95)2o4的红外光谱(ir)图。
图3e是er3+:y3al5o12@ni(fe0.05ga0.95)2o4的红外光谱(ir)图。。
图3f是er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的红外光谱(ir)图。
图4a是在声催化剂er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的存在下磺胺溶液不同时间段的紫外吸收光谱图。
图4b是在催化剂存在与不存在情况下对磺胺溶液的降解效果比较图。
图5a是不同捕获剂对声催化降解磺胺溶液的影响图。
图5b是使用次数对声催化剂er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4声催化降解磺胺溶液活性的影响图。
图6是er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4降解污染物的机理图。
具体实施方式
实施例1新型z型声催化剂er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4
(一)制备方法
1、制备er3+:y3al5o12纳米粉末
将2.7281ger2o3(99.99%)、0.0464gy2o3(99.99%)粉末溶解在浓硝酸(65.00%)中并磁力加热搅拌直至无色透明,得稀土离子溶液。称取12.6208gal(no3)3·9h2o(99.99%)溶解在蒸馏水中,在室温下用玻璃棒搅拌并慢慢加入到稀土离子溶液中。将柠檬酸作为螯合剂和助溶剂,按摩尔比,柠檬酸:稀土离子=3:1,先称取柠檬酸并用蒸馏水溶解后加入到上述混合液中,在50-60℃加热搅拌,当溶液呈粘稠状时停止。在这个过程中没有沉淀生成,最终得到发泡黏胶状溶液。将发泡黏胶状溶液放入烘箱恒温80℃加热36h。在干燥过程中直到蒸干溶剂没有沉淀物生成,最终得到泡沫溶胶。得到的溶胶在500℃加热50min,然后在1100℃煅烧2h。最后,从高温炉中取出烧结的物质,并且在空气中冷却至室温,得到er3+:y3al5o12粉末。
2、制备ni(fe0.05ga0.95)2o4纳米粉末
将10mmolga2o3固体加入到10mmol硝酸镍溶液中,将0.0007mmol的fe(no3)3·9h2o加入到混合液中,产生的混合液用1mol/l氢氧化钠调节ph到12(边调边搅拌30min),得到的悬浮溶液转移到反应釜中180℃下48h,冷却至室温,得到浅蓝色沉淀物,用去离子水清洗数遍,得到的沉淀物在80℃下烘干8小时得到ni(fe0.05ga0.95)2o4粉体,将粉体研细,在500℃的马弗炉中,焙烧2h,取出后再经研磨即得到ni(fe0.05ga0.95)2o4纳米粒子。
3、制备er3+:y3al5o12@ni(fe0.05ga0.95)2o4纳米粉末
将10mmolga2o3固体加入到10mmol硝酸镍溶液中,再加入0.0007mmol的fe(no3)3·9h2o,产生的混合液用1mol/l氢氧化钠调节ph到12(边调边搅拌30min),然后加入4.30mmoler3+:y3al5o12继续搅拌20min。得到的悬浮溶液转移到反应釜中,于180℃下反应48h,冷却至室温,得到浅蓝色沉淀物,用去离子水清洗数遍。得到的沉淀物在80℃下烘干8h,研磨,再于500℃的马弗炉中,焙烧2h,取出后再经研磨,得到er3+:y3al5o12@ni(fe0.05ga0.95)2o4纳米粒子。
4、制备er3+:y3al5o12@ni(fe0.05ga0.95)2o4/au纳米粉末
将10mmoler3+:y3al5o12@ni(fe0.05ga0.95)2o4纳米粉末和7.8mlhaucl4溶液溶解在200ml乙醇中,并利用超声充分分散30分钟(40khz,超声波输出功率为50w),得悬浮液,将悬浮液加热到60-65℃,保持恒温30分钟,过滤,洗涤,分离出来的粉末在350℃下煅烧2h,研磨,得到er3+:y3al5o12@ni(fe0.05ga0.95)2o4/au。
5、制备bivo4纳米粉末
将5mmolbi(no3)3·5h2o完全溶解在硝酸中形成溶液a,5mmolnh4vo3溶解在氢氧化钠中形成溶液b。将溶液b逐滴加入到溶液a中形成黄色悬浮溶液,充分搅拌后,用氢氧化钠调节ph=7,继续搅拌30min,然后转移到反应釜中,180℃,24h后,得产物,清水洗数次,最后干燥,取出后,再经研磨,得bivo4纳米粒子。
6、制备er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4纳米粉末
将10mmoler3+:y3al5o12@ni(fe0.05ga0.95)2o4/au和10mmolbivo4纳米粉末加入到无水乙醇中,超声分散30min,得悬浮液,将悬浮液加热到65℃,保持恒温30分钟,过滤,洗涤,分离出来的粉末在500℃下煅烧2h,研磨,得到er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4纳米粉末。
(二)检测
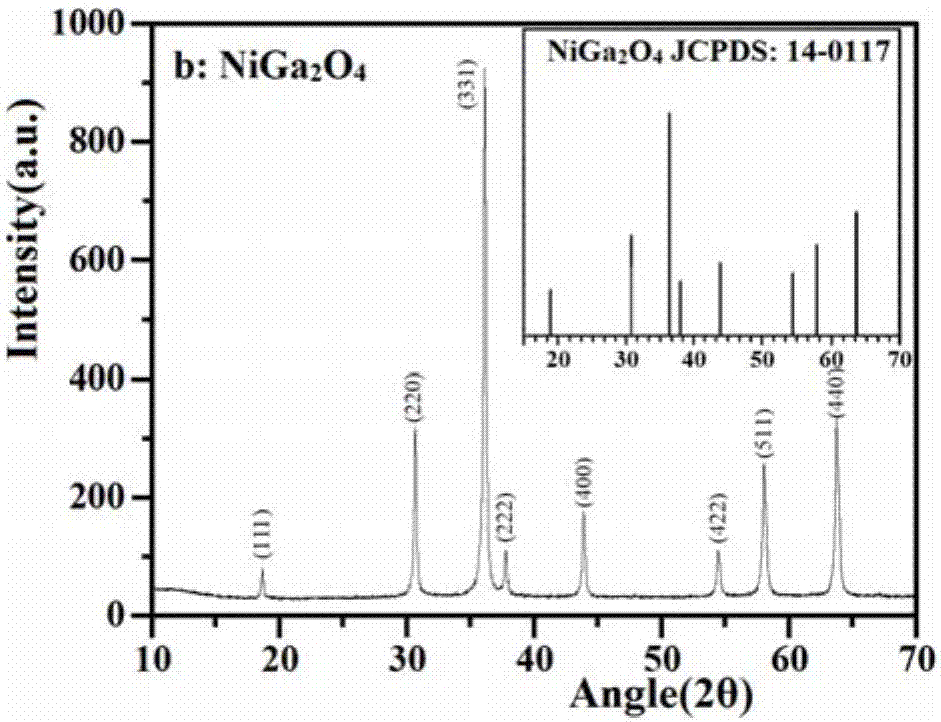
1)制备的样品组成和结构通过xrd来表征,图1a-图1f是er3+:y3al5o12,niga2o4,ni(fe0.05ga0.95)2o4,bivo4,er3+:y3al5o12@niga2o4,er3+:y3al5o12@ni(fe0.05ga0.95)2o4,er3+:y3al5o12@ni(fe0.05ga0.95)2o4-bivo4和er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的x射线粉末衍射(xrd)图。
如图1a,er3+:y3al5o12展示了一些尖的吸收峰在2θ=18.10°(211),27.76°(321),29.78°(400),33.38°(420),35.07°(332),36.68°(422),41.14°(521),46.53°(532),52.74°(444),55.06°(640),57.32°(642)和61.83°(800),这与y3al5o12的标准卡jcpdscard33-0040完全吻合。它证明了er3+:y3al5o12已经被成功制备,而且er3+已经进入了y3al5o12的晶格取代了y3+。
如图1b,制备的试样在2θ=18.6°,30.6°,36.0°,37.7°,43.8°,54.4°,58.0°和63.7°出现尖的吸收峰并且与立方尖晶石结构niga2o4标准卡jcpdscard14-0117的晶面(110),(220),(311),(222),(400),(422),(511)和(440)一一对应。这表明制备的是立方尖晶石结构的niga2o4。
从图1b-1中可以观察到,在2θ=36.0°to2θ=37.7°处的特征峰有轻微的移动它可能是由于fe3+取代了部分ga3+进入了niga2o4晶格当中。
如图1c,制备的bivo4粉末与标准卡jcpdscard14-0688非常吻合。主要的吸收峰出现在15.14°,18.67°,28.58°,30.55°,34.49°,35.22°,40.04°,46.71°,47.31°和54.58°,对应的晶面为(110),(011),(121),(040),(200),(002),(112),(240),(042)和(161)。
在图1d和图d-1中,可以发现除了niga2o4的标准峰以外,er3+:y3al5o12的特征峰也出现,这表明er3+:y3al5o12@niga2o4已经被成功制备。此外,从图1d-1可以注意到在2θ=36.0°到2θ=37.7°处吸收峰有轻微移动,这可以证明er3+:y3al5o12@ni(fe0.05ga0.95)2o4也被成功制备。
在图1e和图1f中,可以发现除了er3+:y3al5o12@ni(fe0.05ga0.95)2o4的特征峰以外,bivo4的特征峰也能被清晰的发现。进一步在图1f中,可以观察到在38.2°和44.4°出现了两个吸收峰,它与au粒子的标准卡jcpds65-2870相符合,对应的晶面分别为(111)和(200)。另外,au粒子的存在可以通过edx进一步来确认。
2)透射电子显微镜(tem)被用来观察样品的详细结构形态,图2a-图2c是er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的透射电子显微镜(tem)图片分析。
图2a-图2c,显示了包覆型复合物er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4在不同放大比例的tem图像,其中,相应的单位比例长度分别为100nm,20nm和10nm。从图2a中可以看出一些片状的微粒是小尺寸的bivo4,其他的大尺寸粒子是包覆了转光剂(er3+:y3al5o12)的ni(fe0.05ga0.95)2o4。图中可以观察到一些微小的黑色粒子分散在大粒子的周围,可以证明au粒子已经被沉积到ni(fe0.05ga0.95)2o4的表面。如图2b所示,放大到20nm的比例尺,可以发现包覆结构更加清晰,从中可以观察到er3+:y3al5o12已经被包覆在ni(fe0.05ga0.95)2o4里面,除此之外一些au纳米粒子嵌入ni(fe0.05ga0.95)2o4和bivo4之间,另一些随机地分散到ni(fe0.05ga0.95)2o4的表面。如图2c,当将单位比例尺进一步放大到10nm,呈现出了许多清晰的晶格条纹。通过计算晶格条纹间距,并且与xrd数据进行对比,制备的声催化剂的晶面可以被确定。他们在图2c中被清晰地标出,如图2c所示,er3+:y3al5o12(d=0.293nm(221)),au(d=0.230nm(111)),ni(fe0.05ga0.95)2o4(d=0.286nm(202))以及bivo4(d=0.315nm(121))。此外,tem的结果表明er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4形成了一个以er3+:y3al5o12为中心的核-壳包覆的结构。
3)图3a-图3f,是er3+:y3al5o12,niga2o4,bivo4,ni(fe0.05ga0.95)2o4,er3+:y3al5o12@ni(fe0.05ga0.95)2o4和er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的红外光谱(ir)图片分析。
图3a呈现了er3+:y3al5o12的红外光谱图。根据早期报道在799.3cm-1和461.6cm-1处是金属和氧之间的振动(y–o和al–o)。观察3b和图3d的红外光谱,可以发现两个强的吸收峰。推断出这些吸收峰是由两个亚点阵形成的,分别为四面体位置和八面体位置的单相的尖晶石结构。在452.5nm处的吸收带主要是由于四面体位置金属氧的拉伸振动引起的;而636.2nm处的吸收带主要归因于八面体配合物造成的这与niga2o4的特征吸收非常吻合。另外从图3d还可以注意到,出现在959.44cm处的吸收峰可能是fe–o伸缩振动,再次证明fe被成功掺杂到niga2o4中。从图3cbivo4的红外光谱中,可以观察到在3439.4nm处的吸收峰可能属于吸附的水分子的伸缩振动;同时,在1636.5nm处的吸收峰很可能是由于吸附的水分子的-oh基团变形振动引起的。另外,在742nm处的吸收峰很可能是由于vo43-不对称的伸缩振动引起的。从图3e和图3f中可以看出,在799.3cm-1,461.6cm-1,452.5cm-1和632.2cm-1处出现了er3+:y3al5o12和ni(fe0.05ga0.95)2o4特征吸收峰。在图3f中进一步可以看到,除了er3+:y3al5o12和ni(fe0.05ga0.95)2o4特征吸收峰外,在745.23cm-1处出现了bivo4的特征吸收,可以表明包覆的复合催化剂er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4被成功制备。
实施例2新型z型声催化剂er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4在声催化降解磺胺中的应用
(一)在声催化剂er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4的存在下磺胺溶液不同时间段的紫外吸收光谱图。
实验条件:200mger3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4和200ml10.0mg/l的磺胺(sa)水溶液。在温度25℃和压力101325pa下,用50w、40khz超声照射300min,间隔为50min。
结果如图4a所示。一般来说当化合物有双键或者共轭双键的时候π→π*电子转移可能发生在相匹配的紫外光的激发下。与此同时,当一些拥有孤对电子的杂原子存在的时候n→π*电子转移可能发生。从图4a紫外吸收光谱图中,可以看出在紫外区域有一个吸收带出现在257nm,这与苯环和含氮键的π→π*和n→π*电子转移相符。这些出现在紫外区的吸收带可以用来评估溶液中剩余磺胺分子并计算其降解率。当超声照射时间达到300分钟时,在257nm处,最大降解率达到95.64%。结果显示当超声照射300分钟的时候,在257nm处的吸收峰几乎完全消失,表明磺胺分子被彻底降解。
(二)催化剂存在与不存在情况下对磺胺溶液降解的影响。
实验条件:取八个锥形瓶分别标记为(a):us,(b):us/bivo4,(c):us/niga2o4,(d)us/ni(fe0.05ga0.95)2o4,(e)us/er3+:y3al5o12@niga2o4,(f)us/er3+:y3al5o12@ni(fe0.05ga0.95)2o4,(g)us/er3+:y3al5o12@ni(fe0.05ga0.95)2o4-bivo4和(h)us/er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4,每个锥形瓶里面分别放入200mg对应的催化剂和200ml10.0mg/l的磺胺(sa)水溶液,(a):us瓶不加入任何催化剂,单纯超声(us)照射。在温度25℃和压力101325pa下,用50w、40khz超声照射300min,记录降解效果。
如图4b所示,(a):us对应于单纯的超声照射,不加入任何催化剂,在300min后降解率仅为38.63%。然而,当加入催化剂的时候,他们的降解率都有提高。尤其是加入bivo4的时候提高明显,这可能是因为bivo4作为窄带半导体可以拓宽光响应范围。从图4b(c-f)中可以看出降解率在逐渐增加,表明上转换发光剂(er3+:y3al5o12)和fe3+的存在扮演了一个重要角色。对于(g)来说,它的降解率达到最高可能是因为ni(fe0.05ga0.95)2o4和bivo4形成了一个z型体系,并且由于au作为导电通道的存在大大提高了电子的传导速率。针对(h),降解率最大,达到95.64%。
(三)不同捕获剂对声催化降解磺胺溶液的影响
为了进一步证实声催化降解磺胺(sa)的反应机理,鉴别反应是否以羟基自由基(·oh)降解为主或者空穴(h+)降解为主是很有必要的。
实验条件:于五个锥形瓶中,分别加入200mger3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4和200ml10.0mg/l的磺胺(sa)水溶液。本实施例选择了四种捕获剂,分别是dmso,叔丁醇(t-buoh),edta和草酸,分别加入四个锥形瓶中,同样也做了一个空白实验,在温度25℃和压力101325pa下,用50w、40khz超声照射300min。在过去,有很多科研工作者已经报道过dmso和t-buoh可以有效地捕获溶液中的·oh。并且众所周知edta和草酸具有捕获空穴的本性。
在图5a中,实验结果显示在不加入任何捕获剂的情况下,磺胺(sa)的降解率在300min时候接近96%。在加入这些捕获剂之后,与空白实验组形成鲜明对照,相比之下磺胺(sa)的声催化降解率均明显降低,其声催化降解率分别下降到54.60%,48.21%,24.88%和30.98%。特别值得注意的是,加入空穴捕获剂的系统里磺胺(sa)的声催化降解率下降的最明显,这可能是由于大部分声催化降解的磺胺(sa)因以bivo4价带上的空穴(h+)氧化为主,因为bivo4具有较高的正电势。dmso和叔丁醇(t-buoh)作为空穴(h+)捕获剂与空穴(h+)发生反应,致使产生某种低反应活性的物质,于是便阻止了声催化降解反应的进行。除此之外,羟基自由基(·oh)氧化也对声催化降解磺胺(sa)做出了一些贡献,作为第二作用。因此可以得出结论,磺胺(sa)的声催化降解是通过空穴(h+)反应和羟基自由基(·oh)反应相结合进行的,但是以空穴氧化为主。
(四)使用次数对er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4降解磺胺性能的影响
实验条件:取1.60ger3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4粉末,分别加入八个锥形瓶,每个锥形瓶里面分别放入200mger3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4和200ml10.0mg/l的磺胺(sa)水溶液。在温度25℃和压力101325pa下,用50w、40khz超声照射300min,记录降解效果。将溶液回收,过滤,收集催化剂,备用。将第一次提取的催化剂烘干煅烧并取出1.20ger3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4粉末,分别加入六个锥形瓶,每个锥形瓶里面分别放入200mger3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4和200ml10.0mg/l的磺胺(sa)水溶液。在温度25℃和压力101325pa下,用50w、40khz超声照射300min,记录降解效果。将溶液回收,过滤,收集催化剂,备用。按上述步骤重复4次。结果如图5b。
由图5b可见,随着重复次数的增加磺胺(sa)降解率略有降低,总体平稳,说明该催化剂稳定可重复使用。
er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4降解磺胺的原理为:niga2o4的带宽3.54ev,其中价带为1.23ev,导带为-2.31ev。bivo4带宽为2.412ev,价带为+2.741ev,导带为0.329ev。由于niga2o4价带电位和bivo4的导带电位相近,因此可以形成较理想的z型光催化体系,可以高效的转移光生电子和空穴对。为了更彻底的转移光生电子和空穴对,本发明将fe3+掺入niga2o4中与bivo4组成一个假定的氧化还原复合中心。在声催化降解反应中,v5+被bivo4导带电子还原成v4+,fe3+被niga2o4价带上的h+自身被氧化成fe4+。此外,fe4+足以在bivo4导带和niga2o4价带形成的假定复合中心氧化v4+,从而再次生成fe3+和v5+重复之前的氧化还原反应。也就是说,声催化降解反应形成了一个er3+:y3al5o12@ni(fe0.05ga0.95)2o4-au-bivo4氧化还原循环体系。值得注意的是,niga2o4导带上的e-和bivo4价带上的空穴被完全分开,并且e-迅速从niga2o4的导带的转移到催化剂au表面,与溶解在水溶液中的氧气分子(o2)反应和生成超氧自由基负离子(·o2-),经过一系列的化学反应成为了羟基自由基(·oh)。这些羟基自由基(·oh)具有较强的氧化能力能够降解周围的有机污染物,生成挥发性副产物或二氧化碳,水和通过矿化产生无机酸。一方面,bivo4价带(vb)上生成的空穴在其结晶粒子的表面可以直接分解有机污染物,直到被完全的分解。另一方面,生成的空穴氧化吸收的水(h2o)分子在bivo4结晶粒子表面产生羟基自由基(·oh),在水溶液中间接降解有机污染物或破坏有机污染物的结构。
- 还没有人留言评论。精彩留言会获得点赞!