一种氨基酸表面活性剂及其制备方法和应用与流程
本发明属于表面活性剂技术领域,特别涉及一种氨基酸表面活性剂、由该氨基酸表面活性剂组成的复配表面活性剂及其制备方法和应用。
背景技术:
氨基酸类表面活性剂是一类以氨基酸为基础的绿色表面活性剂,不仅表面性能优良,同时温和无刺激,亲肤性好,泡沫细腻,抗菌能力强,具有良好的生物降解性和生物相容性等特点,近年来,随着人们对亲肤产品安全意识的提高,氨基酸类表面活性剂越来越多地被应用于高档亲肤品和生物医药等领域。lourdes等采用细胞膜模型研究氨基酸表面活性剂诱导的渗透细胞抵抗力机制,发现了氨基酸表面活性剂具有与细胞膜脂质双层相互作用的特殊能力;pinheiro等阐述了氨基酸表面活性剂与生物界面上的生物分子在细胞层面的相互作用,为以氨基酸表面活性剂为载体的抗微生物药物制剂提供了潜在的医学价值。perinelli等通过制备系列n-酰基氨基酸表面活性剂与不同人体细胞的混合溶剂,考察了其结构与物理化学性质和细胞毒理学特征之间的关系,得出其具有良好的生物相容性。ampatzidis等制备了一系列氨基酸型表面活性剂的o/w型乳液,并采用抛射液滴剖面分析法研究其界面张力,通过流变学研究其表观粘度和稳定性,实验结果显示该类氨基酸表面活性剂乳液能很好地用做化妆品乳液,在化妆品领域中有巨大的潜在应用价值。
目前国外氨基酸表面活性剂已逐步商业化,而国内的相关研究多处于研究阶段,研究成果主要集中在中碳链脂肪酸与谷氨酸、肌氨酸的合成与应用等方面。而中碳链表面活性剂与长碳链表面活性剂相比较,其临界胶束浓度高,应用性能和应用范围受到限制,并且现有氨基酸表面活性剂多以谷氨酸、肌氨酸为原料,产品种类单一,现有提纯工艺多采用传统的石油醚重结晶的提纯方法,产率难以提高,这些都是制约其商业化的主要问题。
技术实现要素:
为了克服上述现有技术的缺点与不足,本发明的首要目的在于提供一种蛋氨酸表面活性剂的制备方法,该表面活性剂通过引入长碳链和蛋氨酸以提高其表面性能,通过利用蛋氨酸含有硫醚键易改性的特性,为其多功能系列产品的开发提供基础;采用了丙酮-水均相反应体系、石油醚重结晶以及无水乙醇萃取的方法制备高产率、高纯度的氨基酸表面活性剂。
本发明另一目的在于提供上述方法制备的蛋氨酸表面活性剂。
本发明另一目的在于提供上述蛋氨酸表面活性剂的应用。
本发明的目的通过下述方案实现:
一种蛋氨酸表面活性剂,为n-脂肪酰基蛋氨酸或n-脂肪酰基蛋氨酸钠;
所述的n-脂肪酰基蛋氨酸的结构式如下所示:
所述的n-脂肪酰基蛋氨酸钠的结构式如下所示:
当n=12时,所述的蛋氨酸表面活性剂为n-肉豆蔻酰基蛋氨酸表面活性剂或n-肉豆蔻酰基蛋氨酸钠表面活性剂;
当n=14时,所述的蛋氨酸表面活性剂为n-棕榈酰基蛋氨酸表面活性剂或n-棕榈酰基蛋氨酸钠表面活性剂;
当n=16时,所述的蛋氨酸表面活性剂为n-硬脂酰基蛋氨酸表面活性剂或n-硬脂酰基蛋氨酸钠表面活性剂。
一种上述的蛋氨酸表面活性剂的制备方法,其主要包括以下步骤:
(1)脂肪酰氯的合成:向反应容器中加入脂肪酸,升温使脂肪酸融化,然后加入催化剂,再边搅拌边滴加socl2,滴加完毕后升温回流反应,反应结束后将所得反应液纯化即得脂肪酰氯;
(2)n-脂肪酰基蛋氨酸的合成:称取蛋氨酸,加入丙酮-水混合溶液,然后滴加naoh溶液至蛋氨酸溶解,同时调节ph=10~11,搅拌至澄清,滴加步骤(1)中得到的脂肪酰氯,在滴加过程中用naoh溶液控制反应液的ph=10~11,滴加完毕反应一段时间,反应结束后将所得反应液纯化即得n-脂肪酰基蛋氨酸表面活性剂;
(3)n-脂肪酰基蛋氨酸钠的合成:称取脂肪酰基蛋氨酸溶于无水乙醇中,再加入naoh溶液,搅拌反应,反应结束后蒸干溶剂即得到n-脂肪酰基蛋氨酸钠表面活性剂。
步骤(1)中所述的脂肪酸优选为棕榈酸、肉豆蔻酸或硬脂酸;所述的升温使脂肪酸融化是指升温至70~80℃;
步骤(1)中所述的催化剂为n,n-二甲基酰胺,所述的催化剂的量为催化量;
步骤(1)中所用的脂肪酸和socl2的摩尔比为1:1~2,优选为1:1.5;所述的滴加是指滴加速度为0.5~1滴/s。
步骤(1)中所述的升温回流反应是指升温至70℃~100℃反应2~5h,优选在90℃反应3h。
步骤(1)中所述的纯化是指将所得反应液减压蒸馏除去剩余的socl2。
步骤(2)中所述的丙酮-水混合溶液是指体积比为1~3:1的丙酮和水组成的混合溶液。
步骤(2)中所述的naoh溶液优选为2mol/l的naoh溶液。
步骤(2)中所述的滴加脂肪酰氯是指滴加速度为0.5~0.8滴/s。
步骤(2)中所用的蛋氨酸和脂肪酰氯的摩尔比为0.8~1.5:0.6~1.2,优选为1.2:1;
步骤(2)中所述的反应一段时间是指在0℃~60℃反应1~6h,优选在30℃反应2.5h。
步骤(2)中所述的纯化指先将反应液减压蒸馏除去溶剂丙酮,然后加入盐酸调节ph=1~2,过滤,将所得固体干燥,再用乙醇溶解,40℃水浴下震荡10min,过滤得到淡黄色澄清溶液,减压蒸馏除去溶剂无水乙醇,再用石油醚重结晶2~3次即得纯化后的n-脂肪酰基蛋氨酸。
步骤(3)中所述的naoh溶液的浓度为0.5~1.2mol/l;
步骤(3)中所述的n-脂肪酰基蛋氨酸和naoh溶液的用量满足n-脂肪酰基蛋氨酸和naoh的摩尔比为1:1。
一种复配表面活性剂,其由椰油酰胺丙基甜菜碱表面活性剂和上述的蛋氨酸表面活性剂复配得到。
优选的,所述的复配表面活性剂指摩尔比为4:6的n-棕榈酰基蛋氨酸表面活性剂和椰油酰胺丙基甜菜碱表面活性剂复配。
上述的n-酰基蛋氨酸表面活性剂和复配表面活性剂在高档化妆品、生物医药、绿色食品等领域中的应用。
本发明的机理为:
本发明采用长碳链脂肪酸和廉价的蛋氨酸为原料合成脂肪酰基蛋氨酸表面活性剂,通过引入长碳链和蛋氨酸以提高其表面性能,通过利用蛋氨酸含有硫醚键易改性的特性,为其多功能系列产品的开发提供基础。在提纯方法方面,采用石油醚重结晶和无水乙醇萃取相结合的提纯工艺使产品产率大幅度提高,为可产业化的低成本合成工艺提供实验依据。而复配表面活性剂中,由于椰油酰胺丙基甜菜碱与本发明的表面活性剂之间形成更稳定的蠕虫胶束体系,从而使复配表面活性剂比单一表面活性剂的表面性能更加优异。
本发明相对于现有技术,具有如下的优点及有益效果:
(1)本发明中采用的蛋氨酸含有硫醚键,易改性,为其多功能系列产品的开发提供基础;
(2)与传统的技术相比,本实验采用丙酮-水为均相反应介质,石油醚重结晶和无水乙醇萃取的提纯方法制备高产率、高纯度的氨基酸表面活性剂。
(3)与采用脂肪酸和谷氨酸、甘氨酸以及肌氨酸合成的氨基酸表面活性剂相比较,本发明具有优异的表面活性,特别是乳化性,本发明的一系列n-酰基蛋氨酸表面活性剂都要优于市面上常售的月桂酰基谷氨酸表面活性剂。
(4)本发明将n-酰基蛋氨酸表面活性剂与椰油酰胺丙基甜菜碱表面活性剂复配使用,得到的复配体系的表面活性比单一表面活性剂的表面性能更加优异。
附图说明
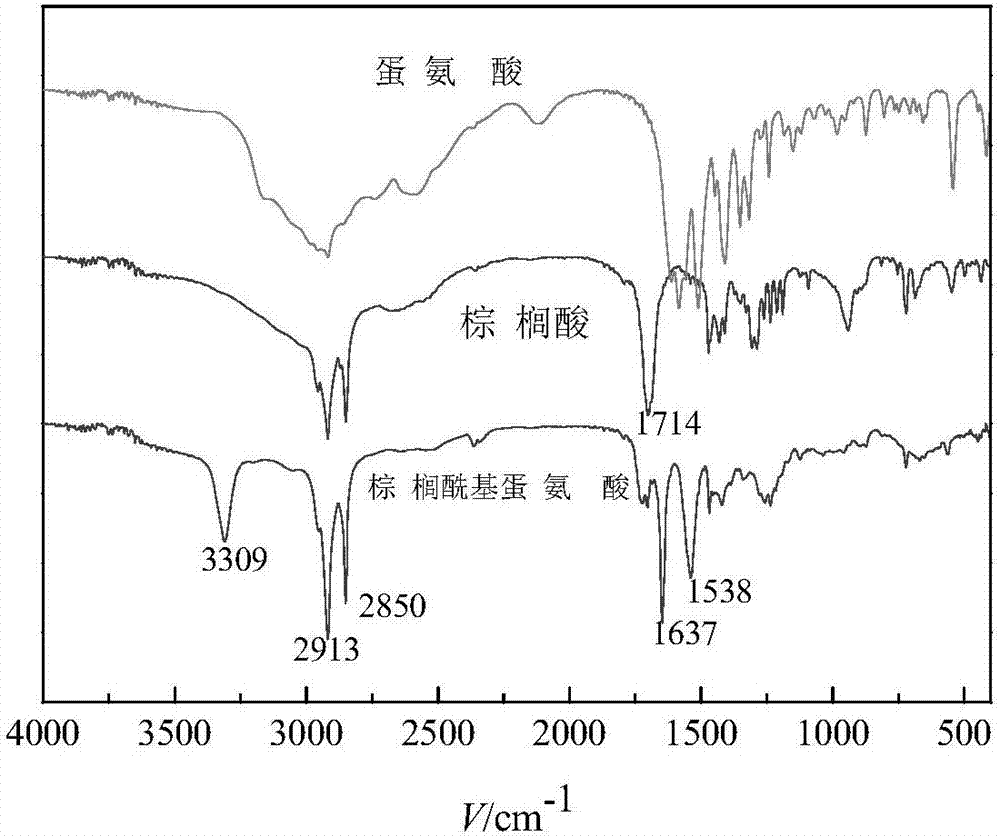
图1为实施例1制备的n-棕榈酰基蛋氨酸的红外光谱图;
图2为实施例1制备的n-棕榈酰基蛋氨酸的核磁共振氢谱图;
图3为实施例1制备的n-棕榈酰基蛋氨酸水溶液在不同浓度下的表面张力图;
图4为实施例2制备的n-肉豆蔻酰基蛋氨酸水溶液在不同浓度下的表面张力图;
图5为实施例3制备的n-硬脂酰基蛋氨酸水溶液在不同浓度下的表面张力图;
图6为实施例2制备的n-肉豆蔻酰基蛋氨酸表面活性剂对苯的増溶能力图;
图7为实施例7中不同摩尔混合比的n-棕榈酰基蛋氨酸和椰油酰胺丙基甜菜碱复配表面活性剂的表面张力图。
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例中所用试剂如无特殊说明均可从市场常规购得。
实施例1
一种氨基酸表面活性剂的制备方法包括以下步骤:
(1)酰氯化反应:在装有冷凝回流的三口烧瓶中加入30.77g棕榈酸,升温到75℃待棕榈酸完全熔化后加入2滴n,n-二甲基酰胺作催化剂,再边搅拌边缓慢滴加13mlsocl2(棕榈酸与氯化亚砜的摩尔比为1:1.5),30min滴加完毕后升温至90℃,继续反应回流3h。收集产物棕榈酰氯,产物性状为暗红色液体,最后在70℃减压蒸馏除去剩余的socl2,得到暗红色的棕榈酰氯,产率为95%。
(2)酰胺化反应:称取7.46gl-蛋氨酸加入带有恒压分液漏斗和温度计的250ml三口烧瓶中,加入15ml去离子水及30ml丙酮(去离子水与丙酮的体积比为1:2),滴加2.00mol/l的naoh溶液至蛋氨酸完全溶解,并且同时调节溶液的ph=10~11;用分液漏斗缓慢滴加11.45g棕榈酰氯,同时用2.00mol/l的naoh溶液调节ph=10~11,滴加完毕后30℃下反应2.5h,收集产物,在40℃减压除去溶剂丙酮;用1.00mol/l的盐酸调节溶液ph=1~2,过滤得到白色固体,干燥除去产物中的水;用适量的无水乙醇溶解产物,在40℃下震荡10min,过滤得到淡黄色澄清溶液,70℃下减压蒸馏除去溶剂无水乙醇,再用适量的石油醚重结晶2~3次得到n-棕榈酰基蛋氨酸表面活性剂,产物呈白色粉末状固体,产率为89%。
(3)盐化反应:称取一定量的棕榈酰基蛋氨酸表面活性剂溶于适量的无水乙醇中,再加入等摩尔量的naoh溶液,50℃下搅拌1h,蒸干溶剂得到n-棕榈酰基蛋氨酸钠。
本实施例制备的n-棕榈酰基蛋氨酸的红外光谱图如图1所示,图1中棕榈酰基蛋氨酸在3309cm-1处是酰胺基团上n-h的伸缩振动吸收峰;2913cm-1和2850cm-1处为亚甲基的伸缩振动吸收峰;1714cm-1处为棕榈酸羧基上c=o的吸收峰,n-棕榈酰基蛋氨酸上此吸收峰消失;1637cm-1和1538cm-1处是酰胺基团上c=o的伸缩振动吸收峰和n-h弯曲振动吸收峰。
本实施例制备的n-棕榈酰基蛋氨酸的核磁共振氢谱如图2所示,1hnmr(500mhz,dmso)δ8.00(s,1h,-nh-),4.25(d,j=12.4hz,1h,-ch-),2.46(s,3h,ch3-s-),2.28–1.92(m,5h,-ch2-c(=o)-nh;-ch2-),1.43(s,2h,ch3-ch2-),1.19(s,20h,-ch2-(ch2)12-),0.80(d,j=7.1hz,3h,-ch3-ch2-)。
图1和图2证明成功合成了目标产物。
实施例2
一种氨基酸表面活性剂的制备方法包括以下步骤:
(1)酰氯化反应:在装有冷凝回流的三口烧瓶中加入29.33g肉豆蔻酸,升温到75℃待肉豆蔻酸完全熔化后加入2滴n,n-二甲基酰胺作催化剂,再边搅拌边缓慢滴加13mlsocl2(肉豆蔻酸与氯化亚砜的摩尔比为1:1.5),30min滴加完毕后升温至90℃,继续反应回流3h。收集产物肉豆蔻酰氯,产物性状为暗红色液体,最后在70℃减压蒸馏除去剩余的socl2,得到暗红色的肉豆蔻酰氯,产率为95.21%。
(2)酰胺化反应:称取7.46gl-蛋氨酸加入带有恒压分液漏斗和温度计的250ml三口烧瓶中,加入15ml去离子水及30ml丙酮,滴加2.00mol/l的naoh溶液至蛋氨酸完全溶解,并且同时调节溶液的ph=10~11;用分液漏斗缓慢滴加10.28g肉豆蔻酰氯(肉豆蔻酰氯与蛋氨酸的摩尔比为1:1.2),同时用2.00mol/l的naoh溶液调节ph=10~11,滴加完毕后30℃下反应3h,收集产物,在40℃减压除去溶剂丙酮;用1.00mol/l的盐酸调节溶液ph=1~2,过滤得到白色固体,干燥除去产物中的水;用适量的无水乙醇溶解产物,在40℃下震荡10min,过滤得到淡黄色澄清溶液,70℃下减压蒸馏除去溶剂无水乙醇,再用适量的石油醚重结晶2~3次得到n-肉豆蔻酰基蛋氨酸表面活性剂,产物呈白色粉末状固体,产率为88%。
(3)盐化反应:称取一定量的肉豆蔻酰基蛋氨酸表面活性剂溶于适量的无水乙醇中,再加入等摩尔量的naoh溶液,50℃下搅拌1h,蒸干溶剂得到n-肉豆蔻酰基蛋氨酸钠。
本实施例制备的n-肉豆蔻酰基蛋氨酸表面活性剂的红外谱图与图1相似,说明本发明也成功制备了目标产物。
实施例3
一种氨基酸表面活性剂的制备方法包括以下步骤:
(1)酰氯化反应:在装有冷凝回流的三口烧瓶中加入34.14g硬脂酸,升温到80℃待硬脂酸完全熔化后加入2滴n,n-二甲基酰胺作催化剂,再边搅拌边缓慢滴加13mlsocl2(硬脂酸与氯化亚砜的摩尔比为1:1.5),30min滴加完毕后升温至90℃,继续反应回流3h。收集产物硬脂酰氯,产物性状为棕红色液体,最后在70℃减压蒸馏除去剩余的socl2,得到棕红色的硬脂酰氯,产率为94.5%。
(2)酰胺化反应:称取7.46gl-蛋氨酸加入带有恒压分液漏斗和温度计的250ml三口烧瓶中,加入15ml去离子水及30ml丙酮,滴加2.00mol/l的naoh溶液至蛋氨酸完全溶解,并且同时调节溶液的ph=10~11;用分液漏斗缓慢滴加12.62g硬脂酰氯(硬脂酰氯与蛋氨酸的摩尔比为1:1.2),同时用2.00mol/l的naoh溶液调节ph=10~11,滴加完毕后30℃下反应2.5h,收集产物,在40℃减压除去溶剂丙酮;用1.00mol/l的盐酸调节溶液ph=1~2,过滤得到白色固体,干燥除去产物中的水;用适量的无水乙醇溶解产物,在40℃下震荡10min,过滤得到淡黄色澄清溶液,70℃下减压蒸馏除去溶剂无水乙醇,再用适量的石油醚重结晶2~3次得到n-硬脂酰基蛋氨酸表面活性剂,产物呈白色粉末状固体,产率为85.32%。
(3)盐化反应:称取一定量的n-硬脂酰基蛋氨酸表面活性剂溶于适量的无水乙醇中,再加入等摩尔量的naoh溶液,50℃下搅拌1h,蒸干溶剂得到n-硬脂酰基蛋氨酸钠。
本实施例制备的n-硬脂酰基蛋氨酸表面活性剂的红外谱图与图1相似,说明本发明也成功制备了目标产物。
实施例4
一种氨基酸表面活性剂的制备方法包括以下步骤:
(1)酰氯化反应:在装有冷凝回流的三口烧瓶中加入30.77g棕榈酸,升温到75℃待棕榈酸完全熔化后加入2滴n,n-二甲基酰胺作催化剂,再边搅拌边缓慢滴加13mlsocl2(棕榈酸与氯化亚砜的摩尔比为1:1.5),30min滴加完毕后升温至90℃,继续反应回流3h。收集产物棕榈酰氯,产物性状为暗红色液体,最后在70℃减压蒸馏除去剩余的socl2,得到暗红色的棕榈酰氯,产率为95%。
(2)酰胺化反应:称取7.46gl-蛋氨酸加入带有恒压分液漏斗和温度计的250ml三口烧瓶中,加入15ml去离子水及15ml丙酮(去离子水与丙酮的体积比为1:1),滴加2.00mol/l的naoh溶液至蛋氨酸完全溶解,并且同时调节溶液的ph=10~11;用分液漏斗缓慢滴加11.45g棕榈酰氯,同时用2.00mol/l的naoh溶液调节ph=10~11,滴加完毕后30℃下反应2.5h,收集产物,在40℃减压除去溶剂丙酮;用1.00mol/l的盐酸调节溶液ph=1~2,过滤得到白色固体,干燥除去产物中的水;用适量的无水乙醇溶解产物,在40℃下震荡10min,过滤得到淡黄色澄清溶液,70℃下减压蒸馏除去溶剂无水乙醇,再用适量的石油醚重结晶2~3次得到棕榈酰基蛋氨酸表面活性剂,产物呈白色粉末状固体,产率为84.15%。
(3)盐化反应:称取一定量的n-棕榈酰基蛋氨酸表面活性剂溶于适量的无水乙醇中,再加入等摩尔量的naoh溶液,50℃下搅拌1h,蒸干溶剂得到n-棕榈酰基蛋氨酸钠。
本实施例制备的n-棕榈酰基蛋氨酸表面活性剂的红外谱图与图1一致,核磁共振氢谱与图2一致,说明本发明也成功制备了目标产物。
实施例5
一种氨基酸表面活性剂的制备方法包括以下步骤:
(1)酰氯化反应:在装有冷凝回流的三口烧瓶中加入30.77g棕榈酸,升温到75℃待棕榈酸完全熔化后加入2滴n,n-二甲基酰胺作催化剂,再边搅拌边缓慢滴加13mlsocl2(棕榈酸与氯化亚砜的摩尔比为1:1.5),30min滴加完毕后升温至90℃,继续反应回流3h。收集产物棕榈酰氯,产物性状为暗红色液体,最后在70℃减压蒸馏除去剩余的socl2,得到暗红色的棕榈酰氯,产率为95%。
(2)酰胺化反应:称取7.46gl-蛋氨酸加入带有恒压分液漏斗和温度计的250ml三口烧瓶中,加入15ml去离子水及45ml丙酮(去离子水与丙酮的体积比为1:3),滴加2.00mol/l的naoh溶液至蛋氨酸完全溶解,并且同时调节溶液的ph=10~11;用分液漏斗缓慢滴加11.45g棕榈酰氯,同时用2.00mol/l的naoh溶液调节ph=10~11,滴加完毕后30℃下反应2.5h,收集产物,在40℃减压除去溶剂丙酮;用1.00mol/l的盐酸调节溶液ph=1~2,过滤得到白色固体,干燥除去产物中的水;用适量的无水乙醇溶解产物,在40℃下震荡10min,过滤得到淡黄色澄清溶液,70℃下减压蒸馏除去溶剂无水乙醇,再用适量的石油醚重结晶2~3次得到n-棕榈酰基蛋氨酸表面活性剂,产物呈白色粉末状固体,产率为80.01%。
(3)盐化反应:称取一定量的n-棕榈酰基蛋氨酸表面活性剂溶于适量的无水乙醇中,再加入等摩尔量的naoh溶液,50℃下搅拌1h,蒸干溶剂得到n-棕榈酰基蛋氨酸钠。
本实施例制备的n-棕榈酰基蛋氨酸表面活性剂的红外谱图与图1一致,核磁共振氢谱与图2一致,说明本发明也成功制备了目标产物。
实施例6
(1)酰氯化反应:在装有冷凝回流的三口烧瓶中加入30.77g棕榈酸,升温到75℃待棕榈酸完全熔化后加入2滴n,n-二甲基酰胺作催化剂,再边搅拌边缓慢滴加13mlsocl2(棕榈酸与氯化亚砜的摩尔比为1:1.5),30min滴加完毕后升温至90℃,继续反应回流3h。收集产物棕榈酰氯,产物性状为暗红色液体,最后在70℃减压蒸馏除去剩余的socl2,得到暗红色的棕榈酰氯,产率为95%。
(2)酰胺化反应:称取7.46gl-蛋氨酸加入带有恒压分液漏斗和温度计的250ml三口烧瓶中,加入15ml去离子水及22.5ml丙酮(去离子水与丙酮的体积比为1:1.5),滴加2.00mol/l的naoh溶液至蛋氨酸完全溶解,并且同时调节溶液的ph=10~11;用分液漏斗缓慢滴加11.45g棕榈酰氯,同时用2.00mol/l的naoh溶液调节ph=10~11,滴加完毕后30℃下反应2.5h,收集产物,在40℃减压除去溶剂丙酮;用1.00mol/l的盐酸调节溶液ph=1~2,过滤得到白色固体,干燥除去产物中的水;用适量的无水乙醇溶解产物,在40℃下震荡10min,过滤得到淡黄色澄清溶液,70℃下减压蒸馏除去溶剂无水乙醇,再用适量的石油醚重结晶2~3次得到n-棕榈酰基蛋氨酸表面活性剂,产物呈白色粉末状固体,产率为84.82%。
(3)盐化反应:称取一定量的n-棕榈酰基蛋氨酸表面活性剂溶于适量的无水乙醇中,再加入等摩尔量的naoh溶液,50℃下搅拌1h,蒸干溶剂得到n-棕榈酰基蛋氨酸钠。
本实施例制备的n-棕榈酰基蛋氨酸表面活性剂的红外谱图与图1一致,核磁共振氢谱与图2一致,说明本发明也成功制备了目标产物。
测试实施例
(1)表面张力测试:配制一定浓度梯度的本实施例1、2和3制备的n-酰基蛋氨酸表面活性剂水溶液,在25℃下测定溶液的表面张力,测试结果分别如图3、4、5所示。
图3为实施例1制备的n-棕榈酰基蛋氨酸表面活性剂水溶液的表面张力图,从图3中可以看出,溶液的表面张力随浓度的升高逐渐降低,最后趋于稳定不变,这时溶液的浓度称为临界胶束浓度,得到n-棕榈酰基表面活性剂的临界胶束浓度为1.2×10-4mol/l,此时表面张力为29.59mn/m,说明此表面活性剂性能较好。
图4为实施例2制备的n-肉豆蔻酰基氨基酸表面活性剂水溶液的表面张力图,从图4中可以看出,溶液的表面张力随浓度的升高逐渐降低,最后趋于稳定不变,这时溶液的浓度称为临界胶束浓度,得到n-肉豆蔻酰基氨基酸表面活性剂的临界胶束浓度为1.25×10-3mol/l,此时表面张力为37.06mn/m,说明此表面活性剂性能较好。
图5为实施例3制备的n-硬脂酰基蛋氨酸表面活性剂水溶液的表面张力图,从图5中可以看出,溶液的表面张力随浓度的升高逐渐降低,最后趋于稳定不变,这时溶液的浓度称为临界胶束浓度,得到n-硬脂酰基蛋氨酸表面活性剂的临界胶束浓度为1.5×10-4mol/l,此时表面张力为22mn/m,表面张力降到20mn/m时浓度c20约为1.4×10-3mol/l说明此表面活性剂性能较好。
(2)乳化性能和泡沫性能测试
乳化性能测试方法:将待测物配制成质量浓度为1g/l的水溶液,取配制好的溶液40ml加入100ml的具塞量筒中,再加入等量的有机溶剂,然后每隔半分钟震荡10次,震荡40次后停止震荡,同时用秒表计时,待析出10ml水时记录时间,重复3次取平均值。结果分别如表1、表2和表3所示。
泡沫性能测试方法:配制150ppm的硬水500ml,待用。用配制好的硬水配制质量浓度为1g/l的表面活性剂溶液,用具塞量筒量取20ml,每隔半分钟震荡10次,震荡40次后停止震荡,记录泡沫高度,同时用秒表计时,分别在3min、5min、10min时记录泡沫高度。
表1n-棕榈酰基氨基酸和月桂酰基谷基酸对不同有机溶剂的乳化时间
表2n-肉豆蔻酰基氨基酸和月桂酰基谷氨酸对不同有机溶剂的乳化时间
表3n-硬脂酰基蛋氨酸和月桂酰基谷氨酸对不同有机溶剂的乳化时间
表4n-肉豆蔻酰基蛋氨酸和月桂酰基谷氨酸的泡沫性能
由表1可知,本实施例1合成的n-棕榈酰基蛋氨酸表面活性剂对五种有机溶剂均有较强的乳化能力,其大小顺序为:苯>甲苯>液体石蜡>三氯化碳>四氯化碳;n-棕榈酰基蛋氨酸的乳化性能力明显高于月桂酰基谷氨酸;
由表2和表3可知,本实施例2合成的n-肉豆蔻酰基蛋氨酸和实施例3合成的n-硬脂酰基蛋氨酸也对二甲苯、甲苯和苯有较强的乳化能力,乳化性能力明显高于月桂酰基谷氨酸。这可能是因为棕榈酰基蛋氨酸的疏水长链比月桂酰基谷氨酸长,胶束聚集数增大且表面张力较小,因此形成的胶束形态较为稳定,乳化能力提高。
由表4可知,本实施例2合成的n-肉豆蔻酰基蛋氨酸的起泡性和泡沫稳定性都优于月桂酰基谷氨酸。
(3)对苯的增溶能力测试
用不同体积的苯增溶在20ml0.001mol/l的蛋氨酸表面活性剂溶液中,采用紫外分光光度法测定本发明的蛋氨酸表面活性剂的增溶能力x(ml/mol),増溶能力x(ml/mol)按下式计算:x=(a×1000)/v×c
式中:a—增溶极限时苯的加入量,ml;v—表面活性剂溶液的体积,ml;c—表面活性剂溶液的浓度,mol/l。
图6为实施例2制备的n-肉豆蔻酰基蛋氨酸表面活性剂对苯的増溶能力图,从图6中可以看出,20ml0.001mol/l蛋氨酸表面活性剂溶液的极限増溶苯的体积为0.12ml,则x(ml/mol)=6000(ml/mol),可以看出n-肉豆蔻酰基蛋氨酸表面活性剂对苯有着优异的吸收能力。
实施例7:复配表面活性剂的制备
将实施例1制备的表面活性剂与椰油酰胺丙基甜菜碱两性表面活性剂(cab)按照摩尔比为0:1、2:8、4:6、6:4、8:2、1:0进行混合,得到不同比例的复配表面活性剂,在25℃,采用吊环法测定不同摩尔比混合的n-棕榈酰基蛋氨酸表面活性剂(pms)和椰油酰胺丙基甜菜碱表面活性剂(cab)复配体系的表面张力,实验结果如图7所示。由图7可知,复配体系的表面活性比单一表面活性剂的表面活性要优异,表现出协同效应。随着椰油酰胺丙基甜菜碱表面活性剂的加入,复配体系的表面活性先提高后降低,在n(pms):n(cab)=4:6时n-棕榈酰基蛋氨酸表面活性剂和椰油酰胺丙基甜菜碱表面活性剂复配表面活性达到最佳,临界胶束浓度达到9.5×10-5mol/l,此时复配体系表面张力为28.1mn/m。这可能是因为随着甜菜碱表面活性剂的加入,其与氨基酸表面活性剂形成了更加稳定的蠕虫胶束体系,复配体系表现出优异的协同作用,当n(pms):n(cab)=4:6时复配体系的表面活性达到最佳,但是随着甜菜碱表面活性剂的加入量的增加,复配体系逐渐显负性,与氨基酸表面活性剂之间的排斥作用增加,使蠕虫胶束体系稳定性下降,其表面活性也略微降低。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!