一种镍基核壳结构催化剂及其制备方法和应用与流程
本发明涉及核壳结构催化剂,具体属于一种页硅酸盐复合氧化物衍生的镍基核壳结构催化剂及其制备方法,以及该催化剂在1,4-丁炔二醇加氢合成1,4-丁二醇的应用。
背景技术:
1,4-丁炔二醇(byd)的氢化是工业上生产1,4-丁二醇(bdo)的主要方法,1,4-丁二醇(bdo)是重要的有机合成和精细化工原料,向下游延伸可生产四氢呋喃(thf)、γ-丁内酯(gbl)、聚氨酯树脂(pu)、聚四亚甲基乙二醇醚(ptmeg)、聚丁二酸丁二醇酯(pbs)等产品,在聚酯新材料、造纸、医药、汽车、农药、纺织和日用化工等领域有广泛而重要的用途。
镍基催化剂由于其廉价性、易得性及高加氢活性,具有极强的工业应用前景,被广泛应用于催化加氢领域。但是,在工业应用过程中,研究者们也意识到了传统镍基催化剂很难满足技术进步对催化剂活性、选择性、稳定性及反应工况的要求。传统负载型催化剂金属纳米颗粒受载体的限制作用较小,临氢水热加氢条件下易于发生活性组分流失,同时伴随各类副反应的发生以及聚合碳物种的结焦。
在过去十年中,核壳结构材料由于其特殊的性质而在众多领域引起了相当大的关注,包括催化,电池,气体传感器,水处理剂,发光,蛋白质分离,药物输送和材料科学。核-壳微粒是由一种纳米材料将另一种纳米材料包覆起来形成的纳米尺度的有序组装结构,其核与壳之间可通过物理或化学作用而相互连接和影响。与负载型纳米催化剂相比,核-壳结构纳米材料具有单分散性、可调变性等优点,不仅能增加活性组分的暴露表面,而且还能通过表面修饰实现对内核或壳层结构、尺寸的控制。
技术实现要素:
本发明的目的在于提供一种用于1,4-丁炔二醇加氢合成1,4-丁二醇的页硅酸盐复合氧化物衍生的镍基核壳结构催化剂及其制备方法,所述的镍基核壳结构催化剂能够有效解决已有镍基催化剂在1,4-丁炔二醇加氢反应中转化率不高,1,4-丁二醇选择性差、收率低,尤其是水热及溶剂热加氢条件下催化剂稳定性差、活性组分易流失和导致结焦等问题。
为实现上述目的,本发明提供的技术方案为:
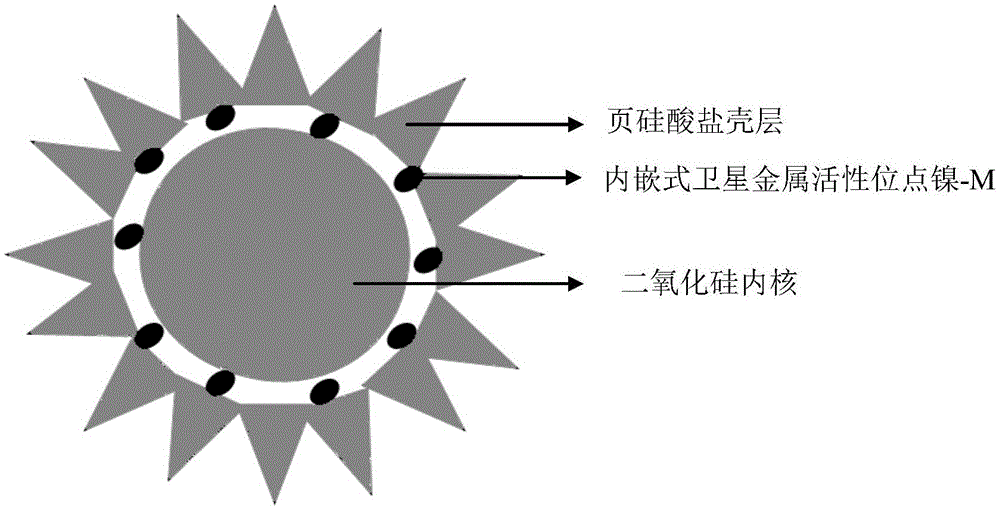
一种适用于1,4-丁炔二醇高选择性液相加氢的页硅酸盐复合氧化物衍生的镍基核壳结构催化剂,由页硅酸盐复合氧化物壳层、内嵌式卫星金属活性位点镍-m和氧化硅内核组成;m为fe,co,cu中的一种(图1)。
所述的页硅酸盐复合氧化物衍生的镍基核壳结构催化剂的制备方法,包括如下步骤:
第一步,将5~25ml浓氨水和50~100ml的体积比1:1~5的水醇溶液于烧杯中混合搅拌10~30min;加入0.1~2ml正硅酸甲酯或正硅酸乙酯,磁力搅拌0.5~6h形成氧化硅晶种;然后在混合溶液中继续加入1~20ml正硅酸乙酯,20~80℃下搅拌1~24h;将混合凝胶乙醇洗涤后离心或过滤,60~140℃干燥8~15h得到sio2球形内核;
第二步,取第一步制得的sio2球形内核1g超声分散于50~200ml的体积比1:1~5的醇水溶液中,并向其中溶解质量比为ni:sio2=0.05~0.35、m:sio2=0.0001~0.15的主金属镍前驱体和辅助金属m前驱体,m为fe,co,cu中的一种,搅拌0.5~3h,然后添加浓氨水至溶液ph=9.5~12,将混合溶液转入水热釜中,100~250℃下水热处理10~50h后,用乙醇或去离子水洗涤至中性。所得固体在80~140℃干燥8~15h,特殊气氛下煅烧,得到镍基核壳结构催化剂。
本发明所述的醇溶剂为甲醇、乙醇、异丙醇中的一种。
本发明中所述主金属镍前驱体为硝酸镍、醋酸镍或氯化镍中的一种;所述辅助金属m前驱体为fe,co或cu相应的硝酸盐、醋酸盐或氯化盐中的一种。
本发明中所述特殊气氛下煅烧方式可选两种,一种为直接在流动氢气氛围下400~600℃煅烧4~10h;另一种为先在400~600℃空气气氛中煅烧2~5h,再切换至流动氢气气氛中煅烧2~5h,本发明涉及煅烧过程,升温速率为1~10℃/min。
所述制得的页硅酸盐衍生的镍基核壳结构催化剂的sio2内核为球型,其直径为200~900nm;所述的壳层为在sio2内核外壁相互作用形成的ni-m双金属页硅酸盐,该壳层为多孔针状或多孔层状,壳层厚度30~80nm;所述的卫星金属活性位点为内嵌于内核和壳层之间的高分散ni-m双金属纳米颗粒,ni为主活性金属,m(fe,co,cu中的一种为辅助活性金属,总的金属含量为5~35wt%。
本发明的镍基核壳结构催化剂适用于原料为含25wt%~35wt%的1,4-丁炔二醇的釜式直接加氢转化。反应条件:溶剂为甲醇、乙醇、异丙醇、二氧六环中的一种,原料体积15~100ml,催化剂用量0.05~0.4g,反应温度25℃~100℃,氢气压力0.5mpa~5mpa,加氢时间1~5h。加氢后的净物料相中1,4-丁炔二醇转化率≥99.9%,1,4-丁二醇收率≥90.0wt%,不饱和生色物质1,4-丁烯二醇(0.1~2wt%)、2-羟基-四氢呋喃(0.1~4wt%)及缩醛2-(4′-羟基丁氧基)-四氢呋喃(0.1~2wt%)等总含量≤8.0wt%。
与现有技术相比,本发明的有益效果:
1.本发明通过简单的方法耦合在氧化硅内核外层形成页硅酸盐。再通过高温还原,在壳层和内核间获得高度分散且均匀排布的金属纳米颗粒。整体镍基核壳结构催化剂具有“页硅酸盐-金属-氧化硅”的“壳层-卫星金属位点-内核”的三层夹心结构。
2.卫星金属镍-m位点来源于起始页硅酸盐复合物中金属组分的还原生长,其点缀于页硅酸盐壳层骨架中,具有很强的金属-载体相互作用,可以分散和限制金属颗粒,大幅提高催化剂于1,4-丁炔二醇加氢反应的活性、目标产物选择性及稳定性。
3.位于高温还原后残余页硅酸盐壳层结构处的配位不饱和的镍离子及辅助金属离子可形成特殊酸性位点,与邻近高分散镍-m卫星位点形成金属-酸双功能活性中心,有利于含缺电子基团的1,4-丁炔二醇吸附和快速转化,促进1,4-丁炔二醇加氢反应。
4.本发明中页硅酸盐壳层的强限域特性能够锚定高活性纳米金属卫星位点,有效地防止活性位点的迁移和高聚物在活性位点的累积,提高催化剂加氢稳定性和抗结焦能力。
5.辅助金属与镍金属电子结构相似,但外层电子或空穴结构不同,可与镍形成双金属协同作用,增强金属表面的氢溢流和氢转移能力,促进材料对氢气的活化,使催化剂具有良好的催化活性和稳定性。
6.由于不同辅助金属的加入在改变金属化学特性的同时,能够改变页硅酸盐壳层的结构和形貌,进可通过不同辅助金属种类和含量调变而可控调节核壳多孔壳层的孔径结构,改善页硅酸盐衍生核壳结构催化剂的孔分布结构及机械强度,使催化剂具有高的加氢活性和循环稳定性。
附图说明
图1由页硅酸盐氧化物壳层,内嵌式卫星金属活性位点镍-m和氧化硅内核组成的三层夹心核壳结构催化剂示意图。
图2为实施例2制得的页硅酸盐氧化物衍生的核壳结构催化剂的透射电镜图(整体)。
图3为实施例2制得的页硅酸盐氧化物衍生的核壳结构催化剂的透射电镜图(局部)。
具体实施方式
本发明不受下述实施例的限制,可根据本发明的技术方案与实际情况来确定具体的实施方式。
实施例1
将5ml浓氨水和50ml水醇溶液(v水:v甲醇=1:3)于烧杯中混合搅拌20min,加入0.1ml正硅酸甲酯,磁力搅拌6h形成球形氧化硅晶种。然后在混合溶液中继续加入5ml正硅酸乙酯,20℃下搅拌1h。将混合凝胶乙醇洗涤后离心,105℃干燥8h得到粒径大小为300nm的sio2球形内核。取制得的sio2球形内核1.0g超声分散于120ml醇水溶液(v异丙醇:v水=1:1)中,并向其中溶解0.4g氯化镍和0.4g氯化钴
镍基核壳结构催化剂1可适用于原料为含35wt%的1,4-丁炔二醇的釜式直接加氢转化:溶剂为二氧六环,原料体积为15ml,催化剂用量0.4g,反应温度50℃,氢气压力1mpa,加氢时间1h。加氢后的净物料相中1,4-丁炔二醇转化率100%,1,4-丁二醇收率94.3wt%,不饱和生色物质1,4-丁烯二醇0.8wt%、2-羟基-四氢呋喃2.4wt%及缩醛2-(4-羟基丁氧基)-四氢呋喃0.6wt%。
实施例2
将25ml浓氨水和80ml水醇溶液(v水:v异丙醇=1:2)于烧杯中混合搅拌30min,加入0.5ml正硅酸乙酯,磁力搅拌1h形成球形氧化硅晶种。然后在混合溶液中继续加入3ml正硅酸乙酯,40℃下搅拌4h。将混合凝胶乙醇洗涤后过滤,90℃干燥10h得到粒径大小为750nm的sio2球形内核。取制得的sio2球形内核1.0g超声分散于160ml醇水溶液(v乙醇:v水=1:3)中,并向其中溶解0.3g硝酸镍和0.06g硝酸铜
镍基核壳结构催化剂2可适用于原料为含25wt%的1,4-丁炔二醇的釜式直接加氢转化;溶剂为甲醇,原料体积为45ml,催化剂用量0.2g,反应温度50℃,氢气压力1mpa,加氢时间5h。加氢后的净物料相中1,4-丁炔二醇转化率100%,1,4-丁二醇收率99.0wt%,不饱和生色物质1,4-丁烯二醇0.1wt%、2-羟基-四氢呋喃0.6wt%及缩醛2-(4′-羟基丁氧基)-四氢呋喃0.1wt%。
实施例3
将10ml浓氨水和85ml水醇溶液(v水:v甲醇=1:1)于烧杯中混合搅拌25min,加入1.8ml正硅酸甲酯,磁力搅拌4h形成球形氧化硅晶种。然后在混合溶液中继续加入1ml正硅酸乙酯,60℃下搅拌16h。将混合凝胶乙醇洗涤后离心,125℃干燥12h得到粒径大小为200nm的sio2球形内核。取制得的sio2球形内核1.0g超声分散于180ml醇水溶液(v异丙醇:v水=1:2)中,并向其中溶解1.2g醋酸镍和0.1g醋酸钴
镍基核壳结构催化剂3可适用于原料为含35wt%的1,4-丁炔二醇的釜式直接加氢转化;溶剂为异丙醇,原料体积为80ml,催化剂用量0.3g反应温度25℃,氢气压力2mpa,加氢时间4h。加氢后的净物料相中1,4-丁炔二醇转化率100%,1,4-丁二醇收率92.7wt%,不饱和生色物质1,4-丁烯二醇1.2wt%、2-羟基-四氢呋喃3.1wt%及缩醛2-(4′-羟基丁氧基)-四氢呋喃0.9wt%。
实施例4
将20ml浓氨水和100ml水醇溶液(v水:v异丙醇=1:5)于烧杯中混合搅拌20min,加入2ml正硅酸乙酯,磁力搅拌5h形成球形氧化硅晶种。然后在混合溶液中继续加入20ml正硅酸乙酯,80℃下搅拌24h。将混合凝胶乙醇洗涤后过滤,140℃干燥15h得到粒径大小为900nm的sio2球形内核。取制得的sio2球形内核1.0g超声分散于200ml醇水溶液(v乙醇:v水=1:4)中,并向其中溶解0.8g硝酸镍和0.04g硝酸铁
镍基核壳结构催化剂4可适用于原料为含30wt%的1,4-丁炔二醇的釜式直接加氢转化;溶剂为乙醇,原料体积为45ml,催化剂用量0.2g反应温度75℃,氢气压力5mpa,加氢时间1h。加氢后的净物料相中1,4-丁炔二醇转化率100%,1,4-丁二醇收率97.6wt%,不饱和生色物质1,4-丁烯二醇0.2wt%、2-羟基-四氢呋喃1.3wt%及缩醛2-(4′-羟基丁氧基)-四氢呋喃0.2wt%。
实施例5
将15ml浓氨水和80ml水醇溶液(v水:v甲醇=1:4)于烧杯中混合搅拌30min,加入1ml正硅酸甲酯,磁力搅拌3h形成球形氧化硅晶种。然后在混合溶液中继续加入13ml正硅酸乙酯,40℃下搅拌8h。将混合凝胶乙醇洗涤后过滤,60℃干燥10h得到粒径大小为700nm的sio2球形内核。取制得的sio2球形内核1.0g超声分散于50ml醇水溶液(v异丙醇:v水=1:5)中,并向其中溶解0.9g氯化镍和0.3g醋酸钴
镍基核壳结构催化剂5可适用于原料为含35wt%的1,4-丁炔二醇的釜式直接加氢转化;溶剂为二氧六环,原料体积为60ml,催化剂用量0.05g反应温度100℃,氢气压力1.5mpa,加氢时间3h。加氢后的净物料相中1,4-丁炔二醇转化率100%,1,4-丁二醇收率96.0wt%,不饱和生色物质1,4-丁烯二醇0.5wt%、2-羟基-四氢呋喃1.9wt%及缩醛2-(4′-羟基丁氧基)-四氢呋喃0.5wt%。
实施例6
将20ml浓氨水和95ml水醇溶液(v水:v异丙醇=1:2)于烧杯中混合搅拌20min,加入1.2ml正硅酸乙酯,磁力搅拌2h形成球形氧化硅晶种。然后在混合溶液中继续加入15ml正硅酸乙酯,20℃下搅拌12h。将混合凝胶乙醇洗涤后离心,125℃干燥8h得到粒径大小为600nm的sio2球形内核。取制得的sio2球形内核1.0g超声分散于80ml醇水溶液(v甲醇:v水=1:2)中,并向其中溶解0.15g醋酸镍和0.004g硝酸铁
镍基核壳结构催化剂6可适用于原料为含25wt%的1,4-丁炔二醇的釜式直接加氢转化;溶剂为甲醇,原料体积为80ml,催化剂用量0.1g,反应温度75℃,氢气压力3mpa,加氢时间4h。加氢后的净物料相中1,4-丁炔二醇转化率100%,1,4-丁二醇收率97.1wt%,不饱和生色物质1,4-丁烯二醇0.3wt%、2-羟基-四氢呋喃1.5wt%及缩醛2-(4′-羟基丁氧基)-四氢呋喃0.3wt%。
实施例7
将25ml浓氨水和50ml水醇溶液(v水:v乙醇=1:3)于烧杯中混合搅拌25min,加入1.5ml正硅酸甲酯,磁力搅拌0.5h形成球形氧化硅晶种。然后在混合溶液中继续加入5ml正硅酸乙酯,60℃下搅拌20h。将混合凝胶乙醇洗涤后离心,90℃干燥12h得到粒径大小为700nm的sio2球形内核。取制得的sio2球形内核1.0g超声分散于90ml醇水溶液(v异丙醇:v水=1:4)中,并向其中溶解1.7g硝酸镍和0.01g氯化铜
镍基核壳结构催化剂7可适用于原料为含35wt%的1,4-丁炔二醇的釜式直接加氢转化;溶剂为乙醇,原料体积为100ml,催化剂用量0.2g,反应温度25℃,氢气压力0.5mpa,加氢时间3h。加氢后的净物料相中1,4-丁炔二醇转化率100%,1,4-丁二醇收率90.0wt%,不饱和生色物质1,4-丁烯二醇2.0wt%、2-羟基-四氢呋喃4.0wt%及缩醛2-(4′-羟基丁氧基)-四氢呋喃2.0wt%。
表1镍基核壳催化剂1~7的反应条件
表2镍基核壳结构催化剂1~7的反应结果
实施例8
取镍基核壳结构催化剂2进行稳定性循环实验,单次釜式直接加氢反应条件同实施例2,催化剂重复使用,无需再活化处理,但每次反应后需要用反应溶剂洗涤,离心后再次与新原料液混合装釜,继续反应。循环反应结果如表3,催化剂在8次循环反应后没有发生活性和收率变化,且催化剂无损失现象,具有较强稳定性。
表3为镍基核壳结构催化剂2循环实验反应结果
- 还没有人留言评论。精彩留言会获得点赞!
- 一种核壳结构Cu-SSZ-13分子筛催化剂及其制备和应用
- 核壳结构多级孔道式钴基费托合成催化剂及其制备方法
- 一种Pt-Au/GR-RuO<sub>2</sub>核壳结构甲醇燃料电池催化剂及应用
- 核壳结构NiCo<sub>2</sub>S<sub>4</sub> @NiCo<sub>2</sub>O<sub>4</sub>纳米针复合催化电极的制备方法
- 一种核壳型铁基金属有机骨架光Fenton催化剂及其制备与应用
- 生产头孢丙烯母核用的催化剂回收装置的制造方法
- 一种核壳状CuAu催化剂及其制备方法和用图
- 一种纳米多孔铜/铂核壳结构催化电极及其制备方法
- 一种燃料电池用核壳型金钴-硼催化剂的制作方法
- 核壳催化剂催化氧化木质素制取芳香基含氧化合物的方法