一种氢解催化剂和制备方法及其在α,α-二甲基苄醇氢解制异丙苯中的应用与流程
一种氢解催化剂和制备方法及其在
α
,
α-二甲基苄醇氢解制异丙苯中的应用
技术领域
[0001]
本发明属于催化技术领域,具体涉及一种氢解催化剂和制备方法及其在α,α-二甲基苄醇氢解制异丙苯中的应用。
背景技术:
[0002]
环氧丙烷(po)是一种重要的有机化工中间体,主要用于生产聚氨酯的原料——聚醚多元醇。目前工业上生产po的工艺主要有氯醇法、共氧化法(po/苯乙烯单体法、po/叔丁醇法、po/甲基叔丁基醚法)、过氧化氢异丙苯法(chp法)和过氧化氢氧化法(hppo法)。日本住友化学(sumitomo)开发的生产po新工艺,包括异丙苯氧化,丙烯环氧化与α,α-二甲基苄醇氢解三个工序。该工艺转化率高与选择性高,产物只有po,不受副产物苯乙烯价格波动的影响,可以为生产商带来更稳定的经济效益。然而,其中α,α-二甲基苄醇氢解工序采用的是cu-cr催化剂,污染环境严重。
[0003]
美国专利us6646139报道了α,α-二甲基苄醇氢解制备异丙苯的工艺技术,该法以cu-cr氧化物为催化剂,其α,α-二甲基苄醇转化率可达100.0%,异丙苯选择性大于97.5%。但该法中的催化剂存在易积炭,cu容易发生高温流失,而cr对环境产生污染等缺点,此外,该类催化剂的使用寿命以及副产物异丙基环己烷含量均没有提及。
[0004]
中国专利cn101735004报道了采用pd或pd-pt金属双功能催化剂,在温度80~160℃,压力0.1~2.0mpa催化α,α-二甲基苄醇氢解制备异丙苯,该法中原料α,α-二甲基苄醇反应前须以碱洗,产生较多废液,且转化率≤99.5%,异丙苯选择性≥99.5%,该催化剂中加氢金属组分采用浸渍法或离子交换法进行负载,负载率低,制造成本高,且存在由于活性过高,易在反应初期引起体系飞温等问题。
[0005]
中国专利cn104151129报道了一种采用ams选择加氢催化剂,由α-甲基苯乙烯制备异丙苯的方法,催化剂由pd/ca/k/mg/ba等组成,以al2o3为载体,α-甲基苯乙烯转化率可达100%,副产物异丙基环己烷可达0ppm,但异丙苯选择性只96%以上,且载体强度不高,催化剂寿命受到限制。
[0006]
中国专利cn1257138c提出了用h2与co混合气还原cu催化剂的方法,由于其所用催化剂仍然是cu-cr催化剂,在该专利中未披露其稳定性的指标。
[0007]
中国专利cn1616383a提出了采用贵金属pd为催化剂,以h2或有机物为氢源,在30~100℃温度下,α,α-二甲基苄醇转化率大于96%,异丙苯选择性大于99%,该反应在间歇式反应釜内进行,实现连续反应有困难。
[0008]
中国专利cn1555348a提出了采用cu基催化剂,使用co含量低于5%的氢气为氢源,该方法能够防止由于氢气压力降低和催化剂中毒引起的活性降低,能有效利用反应容器中的全部容积物,但催化剂仍存在稳定性差的问题。
[0009]
中国专利cn104230642a介绍一种浸渍法制备的pd为催化剂,以h2为氢源,在30~100℃温度下,α,α-二甲基苄醇转化率大于98%,异丙苯选择性大于95%,催化剂存在活性
组分分布不均匀、稳定性不足的风险。
技术实现要素:
[0010]
针对现有技术中存在的上述问题,本发明提供一种氢解催化剂及制备方法,通过将碳源和铝源、硅酸钠、钯源、钇源、助挤剂、水等原料均匀混合制成有一定粘度和流动性的泥状坯料,再通过挤压、干燥、焙烧等过程得到催化剂。本发明制备的氢解催化剂可以应用于α,α-二甲基苄醇氢解制异丙苯,有效解决α,α-二甲基苄醇氢解长周期运行过程催化剂活性组分流失导致活性下降的问题,以及抑制反应副产物的生成,同时提高催化剂的抗液性能。
[0011]
为实现上述发明目的,本发明技术方案如下:
[0012]
本发明提供一种氢解催化剂,以催化剂总质量100%计,包括以下组分:
[0013]
a)pd含量为0.5~3.0%,优选0.5~2.0%;
[0014]
b)y2o3含量为0.5~3.0%,优选1.0~2.0%;
[0015]
c)na2o含量为1.5~13.0%,优选2.0~10.0%;
[0016]
d)sio
2-al2o
3-活性炭含量85~98.0%,优选90.0~95.0%。
[0017]
本发明催化剂中,优选地,所述组分d)sio
2-al2o
3-活性炭,sio2与al2o3、活性炭的质量比范围0.3~8.0:1.0~12:1,优选范围2.0~5.0:1.0~4.0:1。
[0018]
本发明还提供了一种氢解催化剂的制备方法,可用于制备上述氢解催化剂,其步骤包括:
[0019]
将碳源、铝源、硅酸钠、钯源、钇源、助挤剂、水混合,捏合成湿的均匀塑性体,然后挤条成型、干燥、焙烧,得α,α-二甲基苄醇氢解制异丙苯的催化剂。
[0020]
本发明制备方法中,所述钯源、铝源、硅酸钠与碳源的质量比为0.05~6.0:1.0~15.0:1.0~27.3:1,优选1~5:2~10:2~20:1;
[0021]
所述钇源与钯源的质量比为0.1~4.5:1,优选0.5~3.0:1。
[0022]
本发明制备方法中,所述钯源为氯化钯的氨水溶液,其中钯元素的质量浓度为15~25%,优选20%;配制氯化钯的氨水溶液采用的氨水浓度为25~28wt%。
[0023]
所述钇源选自硝酸钇、氯化钇中的一种或两种。金属钯是催化剂的主要活性组分,其含量过高容易造成苯环的过度加氢,降低产品的收率,相反含量过低,催化剂活性偏低,且催化剂失活较快,引入钇助剂能够促进钯的分散,提高催化活性。
[0024]
所述碳源选自本专业熟知的活性碳,可以是椰壳碳、煤制碳以及木质碳等,其粒度为200~1000目之间,碘值为800~1000。活性炭是一种多孔材料,其丰富的孔道结构有助于提高催化剂的活性,并且活性炭具有优异的吸附能力,有利于氢解反应的进行。
[0025]
所述铝源为氧化铝的前驱体,选自伯姆石、拟薄水铝石、sb粉等,优选拟薄水铝石,粒度在200目以下,比表面积在200~500m2/g,孔容在0.3~0.6ml/g。
[0026]
所述硅酸钠为硅酸钠水溶液,固含量≥40wt%,优选为40wt%,模数为1.5~3.0,优选为2.5~3.0。硅酸钠俗称水玻璃,为粘结剂原料并提供助剂na
+
,焙烧过程中当温度升高时(80℃时)水分子重排并对相邻硅醇基之间的缩合起催化作用,进一步加热至120~130℃以上,残存的水分子促使硅醇基缩合,而且si-oh键之间相互脱水缔合,形成si-o-si键,其高温固化后会形成耐水性极好的三维固化结构体系,同时使原料中的钠以及钯离子处于
三维结构的封闭状态中,可以有效防止催化剂长周期运行过程中活性组分钯和助剂钠等的流失,抗液性显著增强。
[0027]
所述助挤剂优选采用田菁粉,加入的助挤剂占原料总质量的1~8%。
[0028]
本发明制备方法中,所述混合,选用的方法包括:先将碳源、铝源、助挤剂进行干混,然后将硅酸钠、钯源、钇源与水(优选去离子水)混合;所述水的用量没有具体要求,使得原料能够形成塑性坯体为基准,在本发明一些实施例中为原料总质量的10~30%;所述干混,电机转速为10~80r/min,时间为30~50min;
[0029]
本发明制备方法中,所述捏合在混捏机中进行;所述捏合,电机转速为10~80r/min,时间为10~30min。
[0030]
本发明制备方法中,所述挤条成型,选用的方法包括:将制备好的均匀塑性体在挤条机中挤出成型,挤条直径为1~5mm;其截面形状可以圆柱形、三叶草、以及四叶草等,挤出速度为20rpm,挤出压力为50~200n。
[0031]
本发明制备方法中,所述干燥优选在空气中进行,干燥温度不超过80℃,优选60~80℃;干燥时间1~24h,优选4~12h。
[0032]
本发明制备方法中,所述焙烧,选用的方法包括:将干燥成型的物料在空气气氛中程序升温焙烧,焙烧条件为:200~400℃下焙烧3~10小时,升温速率为10~100℃/小时。
[0033]
同时,本发明还提供了上述催化剂在α,α-二甲基苄醇氢解制异丙苯中的应用。一种α,α-二甲基苄醇氢解制异丙苯的方法,是以α,α-二甲基苄醇为起始原料,在上面所述的催化剂或上述方法制备得到的催化剂作用下,发生氢解反应制备异丙苯。
[0034]
上述方法中,所述氢解反应,优选在溶剂存在下进行,所述溶剂优选为异丙苯,所述溶剂与α,α-二甲基苄醇质量比范围1:2~4。
[0035]
上述方法中,所述氢解反应,压力为1~2mpa(表压),温度为140~160℃,h2/dmpc(α,α-二甲基苄醇)体积比300~500:1,液时空速1~3h-1
。
[0036]
优选地,本发明所述的催化剂,使用前包括还原活化处理。
[0037]
在一种优选实施方式中,本发明所述的催化剂的还原活化的方法包括:优选首先将反应器温度升至200~230℃,恒温1~2h脱除催化剂吸附的物理水,然后通入含体积分数不超过10v%h2,比如(5v%
±
2v%)h2的氢气和氮气的混合气,对所述催化剂进行预还原至少0.5h,比如1h、1.5h或2h之后逐步提高氢气和氮气混合气中氢气的比例,例如逐步提升至10v%、20v%、50v%、100%,控制该过程催化剂床层热点温度不超过260℃,最后在纯氢气氛下还原2~5h,比如3或4h,还原活化过程保持气体体积空速300~1000h-1
,得到活化的催化剂。
[0038]
本发明方法中,所述原料α,α-二甲基苄醇的转化率为99%以上;异丙苯的选择性为99%以上;催化剂寿命稳定运行达1000h以上,催化剂强度保持良好,且活性组分没有明显流失。
[0039]
本发明的有益效果在于:
[0040]
本发明提供的催化剂,制备过程采用一体化成型方法,以硅酸钠为粘结剂,将粘结剂与浸渍液混合,相比传统浸渍方法,不但节省了催化剂制备步骤,提高了活性组分的分散度,同时解决了催化剂活性组分和助剂钠等流失的问题。其中的氧化铝、氧化硅和活性炭成分主要作为复合载体,兼顾催化剂活性的同时还显著了提高催化剂的硬度、抗液性能等。此
外,催化剂组成中y元素的添加,由于其自身的电负性吸引pd元素形成金属键,可以有效降低钯元素的聚集,提高钯的分散度,进而提高催化剂的抗烧结能力,以及催化剂活性和长周期稳定性。
[0041]
本发明的催化剂用于α,α-二甲基苄醇氢解制异丙苯的反应中,解决了长周期运行过程催化剂活性组分流失导致活性下降,以及抑制反应副产物生成的问题,具有催化剂稳定性好、使用寿命长,原料转化率及产品选择性高等优点。
具体实施方式
[0042]
下面通过具体实施例来说明本发明有益效果。本领域技术人员所应知的是,实施例只用来说明本发明而不是用来限制本发明的范围。实施例中,如无特别说明,所用手段均为本领域常规的手段。
[0043]
一、实施例中主要原料来源:
[0044]
椰壳炭,粒度为200~1000目,碘值为800~1000,购自木林森股份有限公司
[0045]
拟薄水铝石,粒度200目以下,比表面积在200~500m2/g,孔容在0.3~0.6ml/g,购自山东铝业有限公司;
[0046]
sb粉,购自淄博泽浩催化科技有限公司;
[0047]
氯化钯的氨水溶液,配制方法为将氯化钯加入到浓度为25~28wt%的氨水中,然后加入去离子水,使得氯化钯完全溶解并将其中的钯元素含量稀释到质量浓度为20%。
[0048]
如未特别说明,其它原料均为普通市售产品,试剂均为分析纯。
[0049]
二、实施例中产品分析方法:
[0050]
催化剂的侧压强度采用颗粒强度测试仪测定,测定40粒反应后催化剂侧压强度,取其平均值。
[0051]
催化剂中元素含量采用x射线荧光光谱分析(xrf)测定。
[0052]
氢解反应原料中含有的二甲基苄醇的摩尔数、生成的异丙苯的摩尔数以及反应液中残留的二甲基苄醇的摩尔数采用安捷伦7820a气相色谱仪分析后计算,测试条件包括:采用db-5色谱柱、fid检测器,汽化室温度为260℃,检测器温度为260℃,载气为高纯n2、其流速为30ml/min。
[0053]
二甲基苄醇转化率=(1-反应液中残留的二甲基苄醇的摩尔数/原料中含有的二甲基苄醇的摩尔数)*100%;
[0054]
异丙苯选择性=生成的异丙苯的摩尔数/已转化的二甲基苄醇的摩尔数*100%。
[0055]
实施例1:
[0056]
一种氢解催化剂的制备方法,步骤为:
[0057]
1)首先称量70.6g拟薄水铝石、12.5g椰壳炭和3.0g田菁粉在混料机中进行干混,电机转速为15r/min,干混时间为30min,然后将26g去离子水、69.3g硅酸钠水溶液(固含量40wt%,模数约为2.7)、0.87g氯化钇与2.5g氯化钯的氨水溶液(钯元素质量浓度为20wt%)混合,将该溶液加入混捏机中进行捏合,捏合时间为20min,电机转速为15r/min,得到湿料团。
[0058]
3)将步骤2)的湿料团放入双螺杆挤出机中进行挤出成型,挤出的条件为室温,挤出压力70n,挤出速率20rpm,挤出磨具为圆柱型,直径2mm;
[0059]
4)步骤3)所得条状湿基胚体在80℃干燥炉中进行快速干燥8h,最后在马弗炉中速率为60℃/小时升温至400℃下进行焙烧4h,得到催化剂a。
[0060]
实施例1制得的氢解催化剂,由xrf测定其活性组分pd元素含量为0.5wt%,其他组分质量百分含量组成:y2o3含量约为0.5%,氧化钠约为6.5%,其余组分为sio
2-al2o
3-活性炭,sio2与al2o3、活性炭的质量比约为1.6:4.8:1。
[0061]
实施例2
[0062]
一种氢解催化剂的制备方法,步骤为:
[0063]
1)首先称量70.6g sb粉,10.0g椰壳炭和3g田菁粉在混料机中进行干混,电机转速为15r/min,干混时间为30min,然后将20g去离子水、69.4g硅酸钠水溶液(固含量40wt%,模数约为2.7)、1.69g硝酸钇与15g氯化钯的氨水溶液(钯元素质量浓度为20wt%)混合,将该溶液加入混捏机中进行捏合,捏合时间为20min,电机转速为15r/min,得到湿料团。
[0064]
3)将步骤2)的湿料团放入双螺杆挤出机中进行挤出成型,挤出的条件为室温,挤出压力80n,挤出速率20rpm,挤出磨具为圆柱型,直径2mm;
[0065]
4)步骤3)所得条状湿基胚体在80℃干燥炉中进行快速干燥8h,最后在马弗炉中速率为60℃/小时升温至400℃下进行焙烧4h,得到催化剂b。
[0066]
实施例2制得的氢解催化剂,由xrf测定其活性组分pd元素含量为3wt%,其他组分质量百分含量组成:y2o3含量约为0.5%,氧化钠约为6.5%,其余组分为sio
2-al2o
3-活性炭,sio2与al2o3、活性炭的质量比约为2.0:6.0:1。
[0067]
实施例3
[0068]
一种氢解催化剂的制备方法,步骤为:
[0069]
1)首先称量35.3g拟薄水铝石、27.0g椰壳炭和3g田菁粉在混料机中进行干混,电机转速为15r/min,干混时间为30min,然后将22g去离子水、5.2g氯化钇和97.1g硅酸钠水溶液(固含量40wt%,模数约为2.7)与15g氯化钯的氨水溶液(钯元素质量浓度为20wt%)混合,将该溶液加入混捏机中进行捏合,捏合时间为20min,电机转速为15r/min,得到湿料团。
[0070]
3)将步骤2)的湿料团放入双螺杆挤出机中进行挤出成型,挤出的条件为室温,挤出压力70n,挤出速率20rpm,挤出磨具为圆柱型,直径2mm;
[0071]
4)步骤3)所得条状湿基胚体在80℃干燥炉中进行快速干燥8h,最后在马弗炉中速率为60℃/小时升温至350℃下进行焙烧4h,得到催化剂c。
[0072]
实施例3制得的氢解催化剂,由xrf测定其质量百分含量组成为:活性组分pd元素含量3wt%,其他组分质量百分含量组成:y2o3含量约为3%,氧化钠约为9%,其余组分为sio
2-al2o
3-活性炭,sio2与al2o3、活性炭的质量比约为1.03:1.11:1。
[0073]
对比例1
[0074]
1)首先称量58.8g拟薄水铝和3.0g田菁粉在混料机中进行干混,电机转速为15r/min,干混时间为30min,然后将42.5g去离子水、10.2g硝酸钇和114.5g硅酸钠水溶液(固含量40wt%,模数约为2.7)与15g氯化钯的氨水溶液(钯元素质量浓度为20wt%)混合,将该溶液加入混捏机中进行捏合,捏合时间为20min,电机转速为15r/min,得到湿料团。
[0075]
3)将步骤2)的湿料团放入双螺杆挤出机中进行挤出成型,挤出的条件为室温,挤出压力90n,挤出速率20rpm,挤出磨具为圆柱型,直径2mm;
[0076]
4)步骤3)所得条状湿基胚体在80℃干燥炉中进行快速干燥8h,最后在马弗炉中
300℃下进行焙烧4h,得到催化剂d。
[0077]
对比例1制得的氢解催化剂,由xrf测定其质量百分含量组成为:
[0078]
活性组分pd元素含量3wt%,其他组分质量百分含量组成:y2o3含量约为3%,氧化钠约为11%,其余组分为sio
2-al2o3,其含量约为50%氧化铝和33%氧化硅。
[0079]
对比例2
[0080]
1)首先称量46.6g椰壳炭和2.5g田菁粉在混料机中进行干混,电机转速为15r/min,干混时间为30min,然后将138.8g硅酸钠水溶液(固含量40wt%,模数比为2.7)与2.5g氯化钯的氨水溶液(钯元素质量浓度为20wt%)混合,将该溶液加入混捏机中进行捏合,捏合时间为20min,电机转速为15r/min,得到湿料团。
[0081]
3)将步骤2)的湿料团放入双螺杆挤出机中进行挤出成型,挤出的条件为室温,挤出压力100n,挤出速率20rpm,挤出磨具为圆柱型,直径2mm;
[0082]
4)步骤3)所得条状湿基胚体在80℃干燥炉中进行快速干燥8h,最后在马弗炉中300℃下进行焙烧4h,得到催化剂e。
[0083]
对比例2制得的氢解催化剂,由xrf测定其质量百分含量组成为:活性组分pd元素含量0.5wt%,其他组分质量百分含量组成:氧化钠约为13%,其余组分为sio
2-活性炭,sio2与活性炭的质量比约为0.86:1。
[0084]
对比例3
[0085]
1)首先称量47.1g拟薄水铝石、15.3g椰壳炭和2.5g田菁粉在混料机中进行干混,电机转速为15r/min,干混时间为30min,然后将25g去离子水和104g硅酸钠水溶液(固含量40wt%,模数约为2.7)与25g氯化钯的氨水溶液(钯元素质量浓度为20wt%)混合,将该溶液加入混捏机中进行捏合,捏合时间为20min,电机转速为15r/min,得到湿料团。
[0086]
3)将步骤2)的湿料团放入双螺杆挤出机中进行挤出成型,挤出的条件为室温,挤出压力75n,挤出速率20rpm,挤出磨具为圆柱型,直径2mm;
[0087]
4)步骤3)所得条状湿基胚体在80℃干燥炉中进行快速干燥8h,最后在马弗炉中300℃下进行焙烧4h,得到催化剂f。
[0088]
对比例3制得的氢解催化剂,由xrf测定其活性组分pd元素含量5wt%,其他组分质量百分含量组成:氧化钠约为9.7%,其余组分为sio
2-al2o
3-活性炭,sio2与al2o3、活性炭的质量比约为2.0:2.6:1。
[0089]
对比例4
[0090]
1)首先称量58.8g拟薄水铝石、8.8g椰壳炭和2.5g田菁粉在混料机中进行干混,电机转速为15r/min,干混时间为30min,然后将100g氨型硅溶胶(固含量为40%,模数约为2.7)、1.73g氯化钇与2.5g氯化钯的氨水溶液(钯元素质量浓度为20wt%)混合,将该溶液加入混捏机中进行捏合,捏合时间为20min,电机转速为15r/min,得到湿料团。
[0091]
3)将步骤2)的湿料团放入双螺杆挤出机中进行挤出成型,挤出的条件为室温,挤出压力65n,挤出速率20rpm,挤出磨具为圆柱型,直径2mm;
[0092]
4)步骤3)所得条状湿基胚体在80℃干燥炉中进行快速干燥8h,最后在马弗炉中300℃下进行焙烧4h,得到催化剂g。
[0093]
对比例4制得的氢解催化剂,由xrf测定其质量百分含量组成为:活性组分pd元素含量0.5wt%,其他组分质量百分含量组成:y2o3含量约为1%,其余组分为sio
2-al2o
3-活性
炭,sio2与al2o3、活性炭的质量比约为3.4:5.7:1。
[0094]
实施例4-6
[0095]
使用由上述实施例1-3制成的催化剂在用于α,α-二甲基苄醇氢解合成异丙苯工业过程中,催化剂使用前还原活化,方法为:首先将反应器温度升至200~230℃,1.5h脱水,然后通入氢气(5v%)和氮气的混合气预还原0.5h,比如1h,之后逐步提高氢气和氮气混合气中氢气的比例至10v%、20v%、50v%、100%,同时控制催化剂床层热点温度不超过260℃,最后在纯氢气氛下还原3h,还原活化过程保持气体体积空速300~1000h-1
。
[0096]
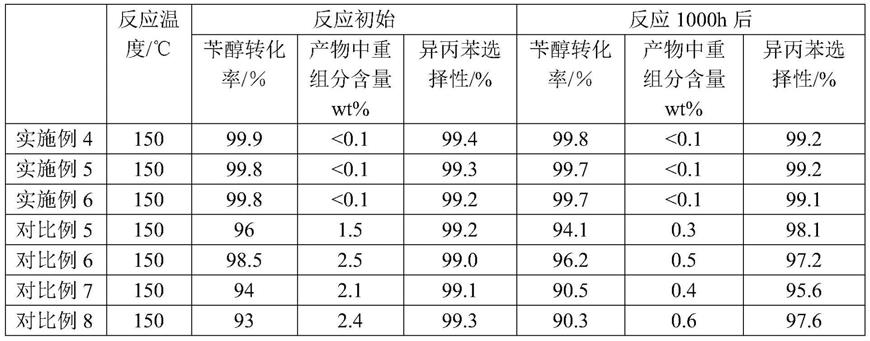
反应过程中原料组成(质量百分含量)为异丙苯75.0%、α,α-二甲基苄醇25.0%,在固定床反应器中,在反应压力为2mpa(表压),h2/苄醇的体积比250:1,原料体积空速为1.5/小时的反应条件下进行1000h考评,反应结果列于表1。对比例5-8:采用对比例1-4制成的催化剂,具体过程同实施例4-6,反应结果列于表1。
[0097]
表1苄醇氢解制异丙苯反应初始及反应1000h后活性评价表
[0098][0099]
由于本发明催化剂中不含cr元素,因此不存在污染环境的问题,从表1中可以看出,α,α-二甲基苄醇在≤160℃,2mpa压力条件下,α,α-二甲基苄醇的转化率>99.0%,异丙苯的选择性>99.0%,该技术在用于α,α-二甲基苄醇氢解合成异丙苯过程中取得了良好的反应结果,采用实施例1制备的催化剂连续运行1000h,苄醇转化率在99%以上,异丙苯选择性在99%以上,且催化剂反应后强度良好,未出现粉化和不完整等缺陷。
[0100]
表2催化剂反应前及反应1000h后强度对照表
[0101]
催化剂反应温度/℃反应前催化剂强度(n/粒)反应后催化剂强度(n/粒)a1505641b1506045c1505846d1505510e1506220f1506035g1505535
[0102]
对实施例及对比例中催化剂反应1000h后进行元素分析,结果如表3所示:
[0103]
表3催化剂反应1000h的pd和na元素分析对照表
[0104][0105]
以上的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通工程技术人员对本发明的技术方案做出的各种变型和改进,均应落入本发明的权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1